the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Sep 2023
| 20 Sep 2023
Technical note: Studying lithium metaborate fluxes and extraction protocols with a new, fully automated in situ cosmogenic 14C processing system at PRIME Lab
Jim Wilson
Allie Koester
Extraction procedures for in situ cosmogenic 14C (in situ 14C) from quartz require quantitative isotopic yields while maintaining scrupulous isolation from atmospheric and organic 14C. These time- and labor-intensive procedures are ripe for automation; unfortunately, our original automated in situ 14C extraction and purification systems, reconfigured and retrofitted from our original systems at the University of Arizona, proved less reliable than hoped. We therefore installed a fully automated stainless-steel system (except for specific borosilicate glass or fused-silica components) incorporating more reliable valves and improved actuator designs, along with a more robust liquid nitrogen distribution system. As with earlier versions, the new system uses a degassed lithium metaborate (LiBO2) flux to dissolve the quartz sample in an ultra-high-purity oxygen atmosphere, after a lower-temperature combustion step to remove atmospheric and organic 14C.
We compared single-use high-purity Al2O3 against reusable 90 %Pt 10 %Rh () sample combustion boats. The boats heat more evenly than the Al2O3, reducing procedural blank levels and variability for a given LiBO2 flux. This lower blank variability also allowed us to trace progressively increasing blanks to specific batches of fluxes from our original manufacturer. Switching to a new manufacturer returned our blanks to consistently low levels on the order of (3.4 ± 0.9) × 104 14C atoms.
We also analyzed the CRONUS-A intercomparison material to investigate sensitivity of extracted 14C concentrations to the temperature and duration of the combustion and extraction steps. Results indicate that 1 h combustion steps at either 500 or 600 ∘C yield results consistent with the consensus value of Jull et al. (2015), while 2 h at 600 ∘C results in loss of ca. 9 % of the high-temperature 14C inventory. Results for 3 h extractions at temperatures ranging from 1050 to 1120 ∘C and 4.5 h at 1000 ∘C yielded similar results that agreed with the nominal value and published results from most laboratories. On the other hand, an extraction for 3 h at 1000 ∘C was judged to be incomplete due to a significantly lower measured concentration. Based on these results, our preferred technique is now combustion for 1 h at 500 ∘C followed by a 3 h extraction at 1050 ∘C. Initial analyses of the CoQtz-N intercomparison material at our lab yielded concentrations ca. 60 % lower than those of CRONUS-A, but more analyses of this material from this and other labs are clearly needed to establish a consensus value.
- Article
(6794 KB) - Full-text XML
- BibTeX
- EndNote





Extracting in situ cosmogenic 14C (in situ 14C) from quartz is challenging in that minute quantities of 14C must be extracted and purified from quartz samples while preventing contamination by ubiquitous atmospheric and organic 14C. These extraction and purification procedures are time-consuming and labor-intensive when done manually – as such they are attractive targets for automation. Lifton et al. (2015) presented results from the initial automated in situ 14C extraction and purification systems at the Purdue Rare Isotope Measurement Laboratory (PRIME Lab), reconfigured and retrofitted from our original glass systems at the University of Arizona. As hoped, the automation of key components of our in situ 14C lab indeed led to increased throughput and reproducibility. While the overall timeline of the extraction, purification, and graphitization was still ca. 3 d, a single person was able to operate these systems simultaneously, boosting sample throughput significantly over the purely manual systems.
These automated systems comprised two independent extraction systems and a separate CO2 purification system. A separate system for converting CO2 to graphite was not automated. This required custom design and implementation of equipment to automate three key aspects of the systems: servo-based valve actuators, temperature control for cryogenic gas purification, and liquid nitrogen (LN) transfer. While these automated systems improved throughput over our original purely manual systems, they also required manual transfer of sample gas between separate extraction, purification, and (manual) graphitization systems.
However, in terms of overall reliability of operation, the limitations of retrofitting our original designs ultimately became apparent. For example, the glass high-vacuum valves are not precision components – no two are precisely the same. The valve actuators thus had to adapt to differences in resistance to motion arising from variations in valve stem and valve bore diameters, as well as to different lengths of travel to adequately seat each valve. As originally designed, the valve actuators accommodated these variations well, but the mechanical settings at which each operated properly tended to creep over time, such that sometimes during active processes individual valves might not indicate that they are closed or open, or might indicate a closed position but not be seated properly and allow leakage across the valve. Without actively checking on the system status when this happened, the sample gas could be pumped away by accident or a process could be interrupted (which could lead to system damage).
Similarly, the LN distribution system in that system was ultimately problematic. LN was transferred from a pressurized 200 L supply dewar through insulated Teflon tubing to fill dewars on various cold traps. Filling and emptying of individual dewars was controlled using LN level sensors comprising three resistors in series, positioned with resistors at empty, nominal, and full levels within each dewar. During the processes, certain cold traps needed to be alternately filled and emptied. Dewars stationed on those traps were emptied using a small shop vacuum cleaner via a drain manifold fitted with cryogenic solenoid valves. Particularly at times of high humidity (not as much of an issue in arid Arizona as in Indiana), ice condensation in those dewars could cause the drain tubing to clog and interrupt the process sequence. In addition, sometimes when a particular dewar was filled and emptied multiple times in a process sequence, the resistor string would not register the proper voltage during a fill cycle to trigger shutoff of LN flow, and the dewar would overflow continuously unless an operator was present to close the main supply dewar valve manually. Thus, although sample throughput and repeatability was considerably improved over manual operation, system reliability was not at the point where one could generally leave a system in unattended operation.
We thus recently purchased and installed a customized Carbon Extraction and Graphitization (CEGS) system from Aeon Laboratories, LLC, similar to that of Goehring et al. (2019) at Tulane University. The largely stainless-steel system (except for specific sections requiring borosilicate glass or fused-silica components) incorporates more reliable valves and improved actuator designs compared to our original system, as well as a robust and efficient liquid nitrogen distribution system (see Goehring et al., 2019, for additional details). The new system, controlled by a flexible and extensible modular software package written in C#, follows a similar procedure to that of Lifton et al. (2015), using a degassed lithium metaborate (LiBO2) flux to dissolve the quartz sample in a research purity (RP) O2 atmosphere. In addition, all sections of the new system are connected, meaning that one can extract all evolved carbon species as CO2 from a quartz sample, purify and precisely measure the resulting gas yield, and convert the CO2 to graphite for AMS analysis – all without human intervention. Below we describe key differences relative to the system of Goehring et al. (2019), and we then present baseline results from the now fully operational system, including procedural blanks and analyses of established intercomparison materials, for both our original single-use high-purity aluminum oxide (Al2O3) and new reusable 90 %Pt 10 %Rh sample boats.
The Purdue CEGS (PCEGS) comprises three main modules: two extraction modules and a collection–purification–graphitization module (main CEGS module) (Fig. 1), following the general design of Goehring et al. (2019) but with an additional extraction module. However, the PCEGS differs from the latter system in two key aspects. First, the two PCEGS extraction modules (Tube Furnace 1, TF1, and Tube Furnace 2, TF2) are connected in parallel, each accommodating a high-temperature resistance furnace with a mullite furnace tube and evacuated by separate vacuum systems distinct from the main CEGS vacuum system (Fig. 1). On the other hand, the Goehring et al. (2019) system comprises one tube furnace in series with the CEGS, evacuated by a single vacuum system. Our design allows each PCEGS extraction module to run processes independently of those controlled by the other modules, enabling increased flexibility in system operations. The other key difference is that condensable gases evolved on the PCEGS during an extraction procedure in either furnace are trapped in a compact borosilicate glass coil trap held at LN temperature (−196 ∘C) instead of the variable temperature trap (VTT) used for this purpose on the Goehring et al. (2019) system (Figs. 1, 2). The compact coil trap (ca. 3.5 cm diameter × ca. 10 cm tall) derives from our previous larger coil trap designs (e.g., Lifton et al., 2001; Pigati et al., 2010; Lifton et al., 2015), which consistently demonstrated quantitative trapping of minute CO2 quantities from O2 carrier gas. This compact design ensures similarly reliable CO2 trapping through a 9 mm o.d. × 7 mm i.d. inlet downtube, delivering process gases directly to the base of the trap before passing through a constriction connecting the downtube to a 6 mm o.d. × 4 mm i.d. coiled section and outlet tube (Fig. 2). The total length of the trap submerged in LN when operating is ca. 55 cm (ca. 5 cm of the downtube and the ca. 50 cm coil).
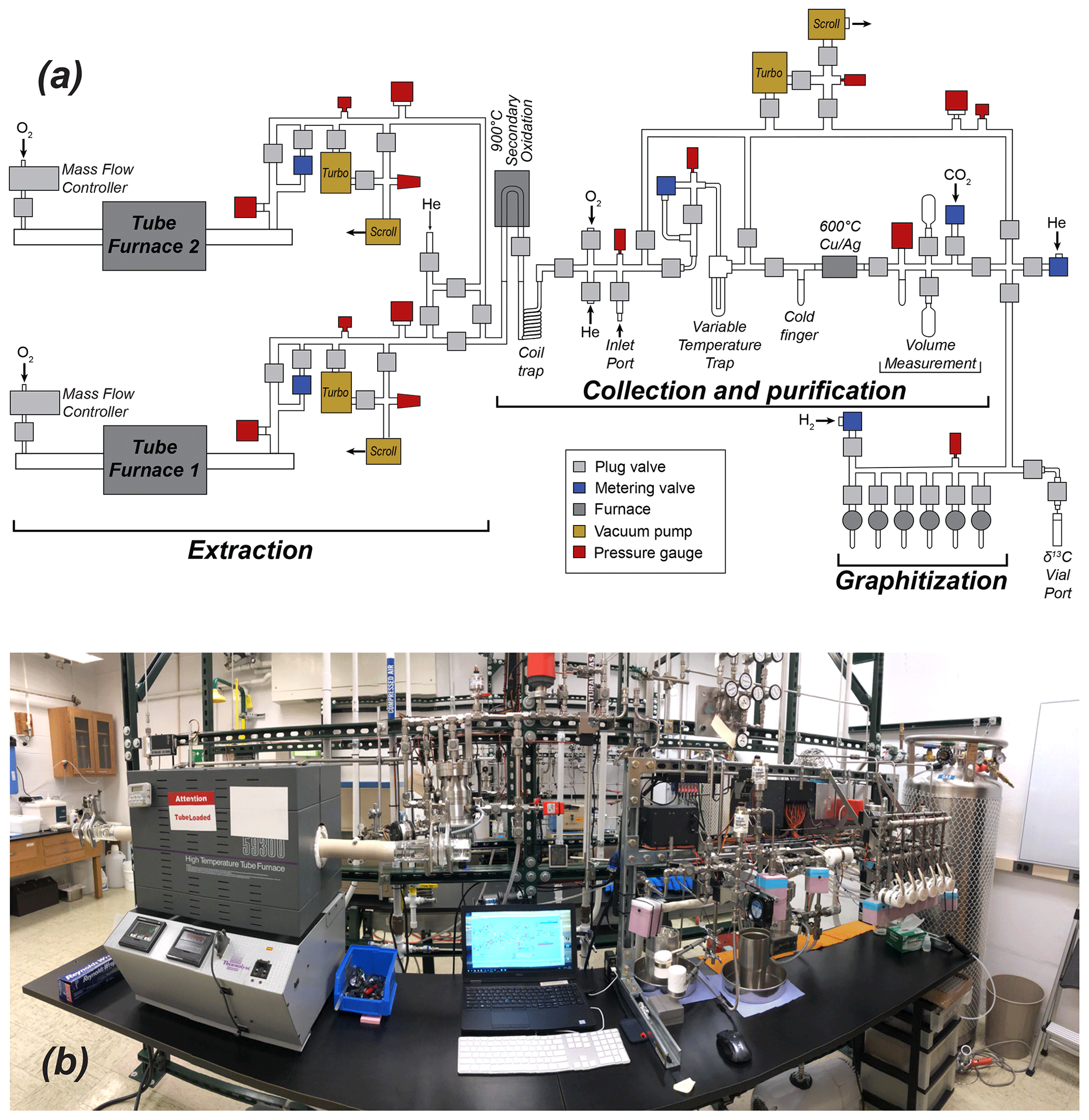
Figure 1(a) Schematic and (b) photo of the Purdue Carbon Extraction and Graphitization System (PCEGS).

Figure 2Compact borosilicate glass coil trap, consisting of a 9 mm o.d. × 7 mm i.d. inlet downtube (on the left), connected to a tightly coiled 6 mm o.d. × 4 mm i.d. section with subsequent outlet tube. The scale on bottom is in centimeters.
Other than incorporating a U-shaped secondary oxidation furnace (9 mm o.d. × 7 mm i.d. filled with 2 mm quartz beads, held at ca. 900 ∘C) from the Lifton et al. (2015) system instead of an inline granular quartz oxidation furnace of the Goehring et al. (2019) design, the rest of the PCEGS utilizes similar hardware to that of the latter. However, our six-reactor graphitization manifold is configured in front of the main purification and measurement process path to achieve a shorter footprint than the linear configuration of the Tulane system (Fig. 1), allowing the first extraction module and the CEGS module to fit onto our existing lab frame and benchtop.
We implement a 2 d extraction procedure with the PCEGS similar to those of Lifton et al. (2015) and Goehring et al. (2019), utilizing a LiBO2 flux to dissolve the quartz and release the in situ 14C at 1100 ∘C (Table 1). The first day's procedures involve degassing the LiBO2 flux and preparing the purified quartz for extraction, while the second day is the extraction, purification, and graphitization procedure. Once started, the day 1 LiBO2 degassing process operates on the selected extraction module (either TF1 or TF2) completely independently of the main CEGS module. The day 2 process, on the other hand, requires control from the main CEGS module to allow sample collection, purification, measurement, dilution, and graphitization. In practical terms, we execute a day 1 process on one extraction module, then the next day start a day 1 process on the second extraction module. The day 2 process for the first extraction module can then be run without interruption from the main CEGS module. The day 1 and day 2 processes are then subsequently cycled between the two extraction modules. This comfortably allows for PCEGS throughput of 4–5 samples per week.
Table 1Procedural flow for in situ 14C extraction at PRIME Lab. See text for abbreviations.

On day 1, a quartz sample is pretreated with 50 % (v : v) HNO3 : 18 MΩ water for at least 90 min in an ultrasonic bath, rinsed thoroughly in 18 MΩ water, then dried in a vacuum oven overnight. A sample boat (either single-use high-purity Al2O3 or reusable 90 %Pt-10 %Rh) containing ca. 20 g of pre-fused LiBO2 beads (melting point 845 ∘C) is placed inside a flame-cleaned fused-silica sleeve in the mullite furnace tube (with borosilicate glass o-ring ball joint end seals) using flame-cleaned implements. The 24 in. long (60.96 cm) fused-silica sleeve (replaced after every sample) extends beyond the furnace hot zone, protecting the furnace tube from LiBO2 vapors that evolve from the fused sample at high temperature. The aggressively reactive vapors etch the interior of the sleeve within the hot zone of the furnace instead of the furnace tube itself (Fig. 3). To minimize intrusion of atmospheric CO2 or other contaminant gases into the furnace tube each time it is opened, the tube is first backfilled with research purity He (99.9999 %) to 20 torr above ambient atmospheric pressure. The He is then slowly bled through the tube while open to atmosphere. Once closed again, the furnace tube is evacuated to torr, isolated, and 50 torr of RP O2 is subsequently added. The furnace is then heated to the extraction temperature (typically 1100 ∘C) for 1 h while O2 is bled through with a mass flow controller and automated metering valve to maintain the tube pressure and to flush out any evolved contaminants to the vacuum pump. The tube is then cooled to <800 ∘C to ensure LiBO2 solidification before evacuation overnight.

Figure 3Comparison of quartz sleeve corrosion from LiBO2 for (a) Al2O3 boats and (b) boats (inset) after the high-temperature fusion step (3 h) at the temperatures indicated. Note the significantly greater corrosion associated with the boats vs. the Al2O3, indicating more aggressive and uniform heating in the former and noticeably milder corrosion from the 1050 and 1000 ∘C runs. The Al2O3 boat on the left side of (b) is holding the sleeves in place but also serves as a comparison to (a). We speculate that the greater corrosion from LiBO2 in the boats reflects higher thermal conductivity of the metal boats vs. the sintered ceramic boats.
On day 2, after backfilling the tube with He as before, the boat with degassed LiBO2 is removed (again with flame-cleaned implements) to a HEPA-filtered laminar flow bench. Approximately 5 g of the pretreated quartz sample is evenly distributed over the now-solid LiBO2 in the boat and the boat and sample are returned to the furnace, evacuated to torr, isolated, and 50 torr RP O2 is added again. The sample is then heated to 500 ∘C for 1 h to combust and remove atmospheric and organic contaminants, while bleeding O2 across the sample as before and exhausting to the vacuum system. After that hour, the 500 ∘C tube furnace is evacuated to torr. Subsequently, 50 torr of RP O2 is admitted into the tube furnace and the sample and flux are heated to 1100 ∘C and held at the high temperature for 3 h to dissolve the quartz and release any trapped carbon species. During extraction, the O2 pressure in the tube typically rises to ca. 60 torr.
After the extraction procedure completes, the evolved gases are bled with RP O2 through the secondary oxidation furnace to ensure any carbon species released during extraction are completely oxidized to CO2 before collection in the compact coil trap cooled with LN. During this step, the tube pressure at the end of the extraction step is maintained during the bleed (to prevent excess LiBO2 vaporization) while the furnace cools to <800 ∘C (to ensure complete melt solidification), before shutting off additional O2 inflow and slowly evacuating all tube gases through the secondary furnace and coil trap. The condensed gases are then transferred to the purification section to remove water, halogens, and nitrogen and sulfur oxides. The gas is transferred cryogenically with LN first into the variable temperature trap (VTT), and the incondensable gases are evacuated. The VTT is then warmed to −145 ∘C for 10 min to remove sulfur oxides, water, and other contaminant gases from the sample CO2. The evolved CO2 is then passed through a Cu mesh Ag wool trap held at 600 ∘C (removes nitrogen oxide, halogen, and sulfur oxide contaminants) and frozen with LN into the volumetric measurement chamber (MC) (Fig. 1). The CO2 yield is then measured manometrically as equivalent mass of C (µg) and typically diluted to ca. 300 µg C with 14C-free CO2. If the undiluted CO2 yield is ≥300 µg C, no dilution step occurs. A ca. 9 µg C split (ca. 3 % of the total C mass) is collected in a pre-evacuated Exetainer® vial for stable C isotopic analysis offline, and the remaining sample is transferred cryogenically to one of the six graphite reactors (Fig. 1). The sample then undergoes hydrogen reduction (with research purity H2 – 99.9999 %) to filamentous C (graphite) on an Fe catalyst, with water trapped by Mg(ClO4)2 (Southon, 2007; Santos et al., 2004). Procedural background samples are run after approximately every 7–10 unknown samples, using identical procedures without adding quartz.
Finally, the graphite is packed into an Al cathode for 14C measurement by accelerator mass spectrometry (AMS) at PRIME Lab. Sample ratios are measured relative to Oxalic Acid II (NIST-4990C). Stable carbon isotopic ratios were measured at the University of California at Davis Stable Isotope Facility (https://stableisotopefacility.ucdavis.edu, last access: August 2023) using isotope ratio mass spectrometry (Lifton et al., 2015). Measured in situ 14C concentrations are calculated from the resulting after subtracting representative procedural background 14C values, following Hippe and Lifton (2014). Measurement uncertainties are presented at the 1σ level unless otherwise noted.
Once the PCEGS was operational, we began to characterize its performance in terms of procedural blank (background) values and measurements of intercomparison materials such as CRONUS-A (Jull et al., 2015). We also characterized the mass dependence of graphitization blanks. Since the publication of Lifton et al. (2001), we and other labs using LiBO2 for extraction (e.g., Goehring et al., 2019; Lamp et al., 2019; Fülöp et al., 2010) had used single-use high-purity sintered Al2O3 combustion boats for our flux + samples. On the other hand, laboratories that implemented flux-free in situ 14C extractions have either used Pt (e.g., Hippe et al., 2009, 2013; Lupker et al., 2019) or fused-silica vessels (Fülöp et al., 2015, 2019). The labs using flux-free processes typically report blanks on the order of 1–3 × 104 14C atoms (e.g., Lupker et al., 2019; Fülöp et al., 2019), while the labs using flux-based extractions have reported blanks on the order of 1–2 × 105 14C atoms (e.g., Lifton et al., 2015; Goehring et al., 2019; Lamp et al., 2019). Goehring et al. (2019) deduced that the differences in 14C backgrounds between the flux and flux-free extraction systems lay at least in part with the sintered Al2O3 boats reacting with the flux to release small and variable amounts of persistent contaminant 14C during the extraction process. They described assessing boats of alternate construction and reported a promising process blank result of ca. 4×104 14C atoms from an initial experiment with a reusable 90 %Pt : 10 %Rh alloy boat.
Our initial PCEGS experiments utilized the single-use Al2O3 combustion boats, but in the meantime we also obtained a set of 90 %Pt : 10 %Rh (hereafter ) combustion boats from Heraeus Precious Metals North America LLC (http://www.pt-labware.com, last access: August 2023). We thus compared results using both types of boats for both blanks and intercomparison samples. The solidified LiBO2 + sample melt is cleaned from the boats between samples by overnight ultrasonication at 40 ∘C in 10 % (v : v) reagent grade HNO3 : 18 MΩ water in sealed 1 L polypropylene bottles, followed by thorough rinsing in 18 MΩ water and drying in a gravity oven.
3.1 Graphitization blanks
The mass dependence of the PCEGS graphitization blanks was assessed by graphitizing aliquots of 14C-free CO2 in masses ranging from ca. 50 to 1000 µg C (Table 2). As with previous studies using either Zn or H2 as the reducing agent for CO2 to C (e.g., Donahue et al., 1990; Lifton et al., 2001, 2015; Goehring et al., 2019), we observe an inverse relationship between sample mass (in µg C) and the measured . If one assumes a constant modern contaminant contribution to the graphitization blank from the graphitization reactor, independent of the sample mass, then one would observe an inverse mass dependence of the blank: higher blanks for lower-mass samples (e.g., Donahue et al., 1990). This relationship is well-characterized by the following equation (adjusted R2=0.994):
Correction of the measured sample for the graphitization blank (Bg) follows Eq. (6) of Donahue et al. (1997).
Table 2Graphitization blanks.
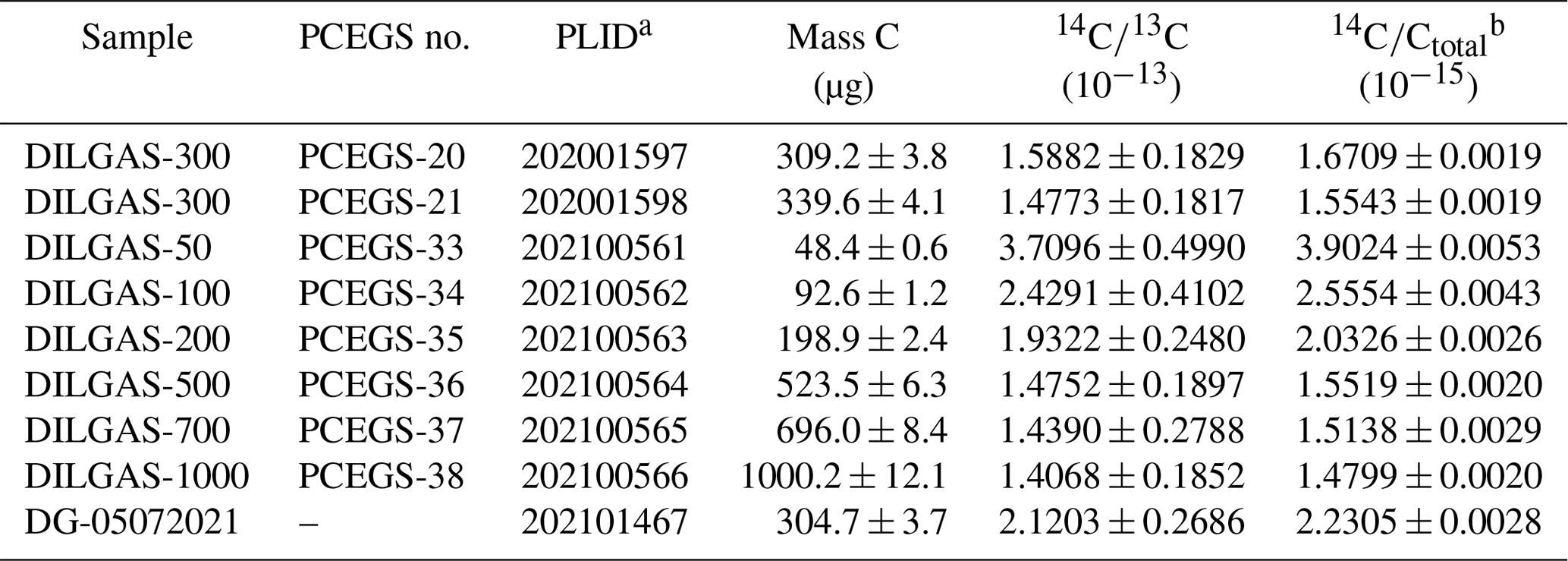
a PRIME Lab ID. b δ13C averages −45.6 ± 0.2 ‰VPDB.
3.2 Procedural blank comparison
Initial experiments with the new system involved procedural blanks with our original single-use Al2O3 boats in concert with measurements of intercomparison materials (Sect. 3.3). Subsequently, we switched to reusable 90 %Pt 10 %Rh sample boats, with associated measurements of procedural blanks and intercomparison materials for a range of experimental conditions.
3.2.1 Al2O3 boats
The first set of blanks and intercomparison samples processed on the PCEGS with Al2O3 boats involved a more aggressive than normal day 2 combustion step to more thoroughly remove any potential organic C that might remain on the etched sample grains. This was motivated by Nichols and Goehring (2019), who found evidence of modern 14C contamination by laurylamine used in froth flotation mineral separation techniques that was not removed completely by their original etching procedure. Although we had not observed evidence of this issue with in situ 14C results from our lab, we tested a low-temperature combustion procedure of 2 h at 600 ∘C, reasoning that Hippe et al. (2013) utilized a 2 h at 700 ∘C combustion step with no apparent demonstrable effects on their results relative to combustion for 1 h at 500 ∘C. This more aggressive combustion step was then followed by our normal 1100 ∘C flux fusion for 3 h.
Initial procedural blank experiments largely utilized TF1 and progressively increased from ca. 6.50×104 to 1.03×105 14C atoms with a mean of (8.79 ± 1.64) × 104 14C atoms, while a single blank from TF2 yielded ca. 1.14×105 14C atoms (Table 3, Fig. 4). The source of the time-dependent increase was not identified before switching to the boats, but these values still represent an improvement over blank values presented in Lifton et al. (2015) of ca. 30 %–70 %.
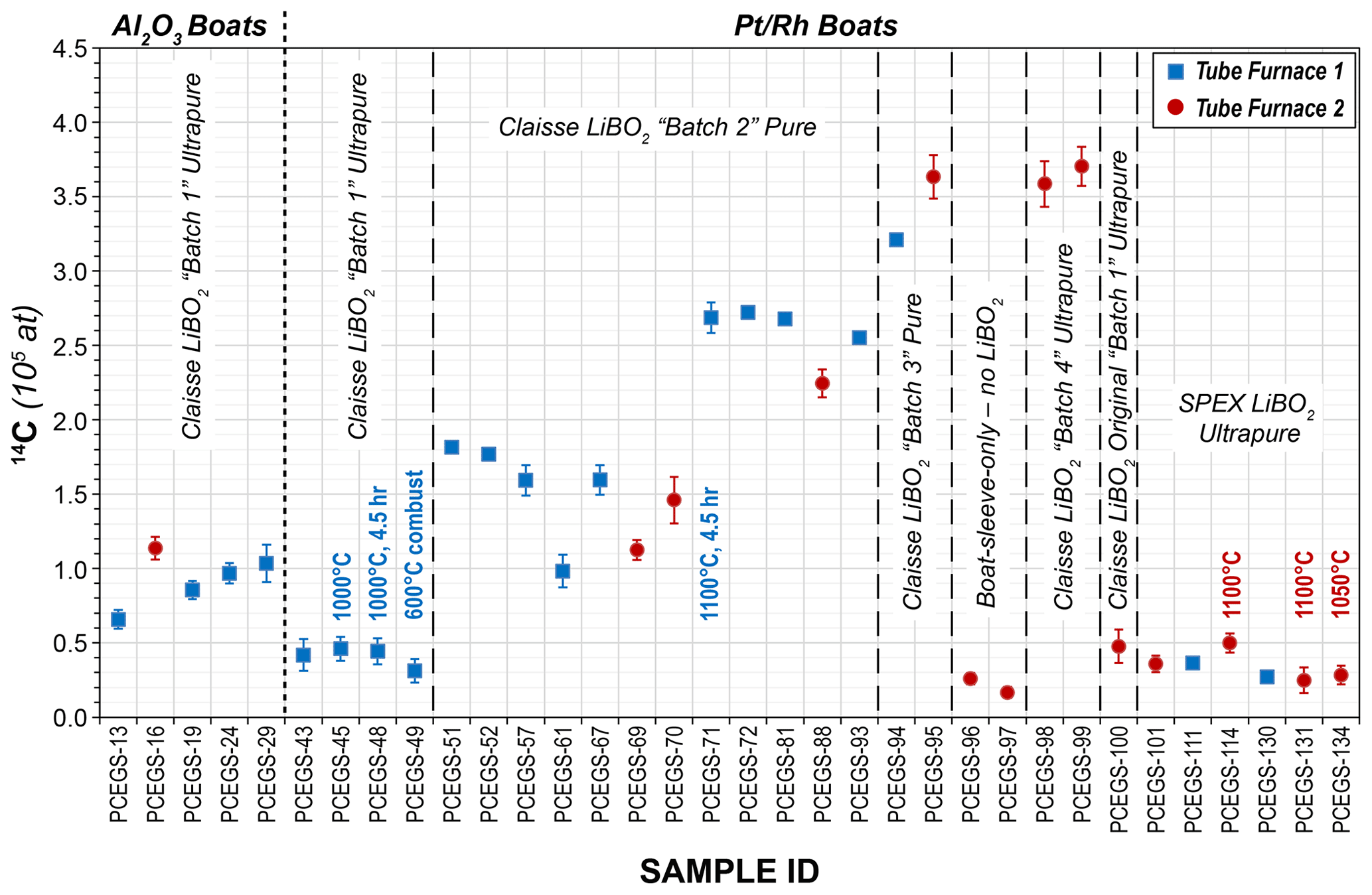
Figure 4Procedural blank results for Al2O3 and boats (1σ uncertainties). All blanks using Al2O3 boats used a 2 h at 600 ∘C combustion step followed by a 3 h extraction at 1100 ∘C (1120 ∘C for Tube Furnace 2 due to a miscalibration at the 1100 ∘C set point). All Tube Furnace 1 runs were a 1 h at 500 ∘C combustion step followed by a 3 h extraction step at 1100 ∘C (except as indicated). Tube Furnace 2 combustions with boats were also 1 h at 500 ∘C, but extractions were at 1120 ∘C due to the miscalibration (except as indicated).
Table 3Al2O3 procedural blanks. All analyses used 2 h combustion at 600 ∘C and 3 h extraction at 1100 ∘C (unless otherwise noted).
All blanks here used Claisse C-0611-00, Batch C-10001 – “Batch 1” ultrapure LiBO2. * 1120 ∘C extraction due to furnace miscalibration at 1100 ∘C set point.
3.2.2 Pt Rh boats
On switching to the boats, we also reverted to our original procedure utilizing a 500 ∘C combustion step for 1 h. It was immediately obvious that the boats heat much more uniformly than the Al2O3 based on dramatic differences in the flux's corrosive effects on the quartz sleeves between the two types of boats (Fig. 3). The sleeves used with the Al2O3 boats were corroded mainly above and below the boat and at the ends of the heated zone where the LiBO2 vapor condenses in ca. 5 cm wide bands (Fig. 3a). The rest of the heated portion of the sleeve is only lightly corroded and remains transparent. However, when using the boats, the LiBO2 more evenly corrodes the sleeve interior over the entire hot-zone length (Fig. 3b). Most of the boat heating likely occurs via conduction from the bottom edges of the boat in contact with the quartz sleeve. It thus appears that the more efficient heat conduction of the metal boats leads to more even and aggressive heating of the flux and sample than in the Al2O3 boats. Experiments with the boats at extraction temperatures of 1000 and 1050 ∘C resulted in significantly less corrosion of the sleeve than at 1100 ∘C (Fig. 3b).
Initial procedural blanks using the boats were dramatically lower than those using the Al2O3 boats, with much better reproducibility, averaging (4.08 ± 0.66) × 104 14C atoms (1σ) (Table 4, Fig. 4). Different combinations of combustion (500 and 600 ∘C for 1 h) and extraction temperatures and times (1100 ∘C for 3 h; 1000 ∘C for 3 and 4.5 h) were investigated as well (Table 4) (corresponding to intercomparison experiments described in Sect. 3.3), with no significant effect on blank results. This supports the hypothesis of Goehring et al. (2019) that a significant component of Al2O3 procedural blanks derived from the sintered ceramic boats themselves. The improved blank reproducibility using the boats allows us to identify background signals that previously we were unable to resolve. After this initial set of analyses depleted most of the bottle of ultrapure-grade LiBO2 (Claisse C-0611-00, Batch C-10001 – “Batch 1”), we switched to a new bottle of pure-grade LiBO2 (Claisse C-0610-00, Batch C-17000-10 – “Batch 2”). We reasoned that pure and ultrapure grades only differ in metal impurity content – both are pre-fused, spherical beads and thus should be essentially functionally equivalent for our application.
Table 4 procedural blanks. All analyses used 1 h combustion at 500 ∘C and 3 h extraction at 1100 ∘C (unless otherwise noted).
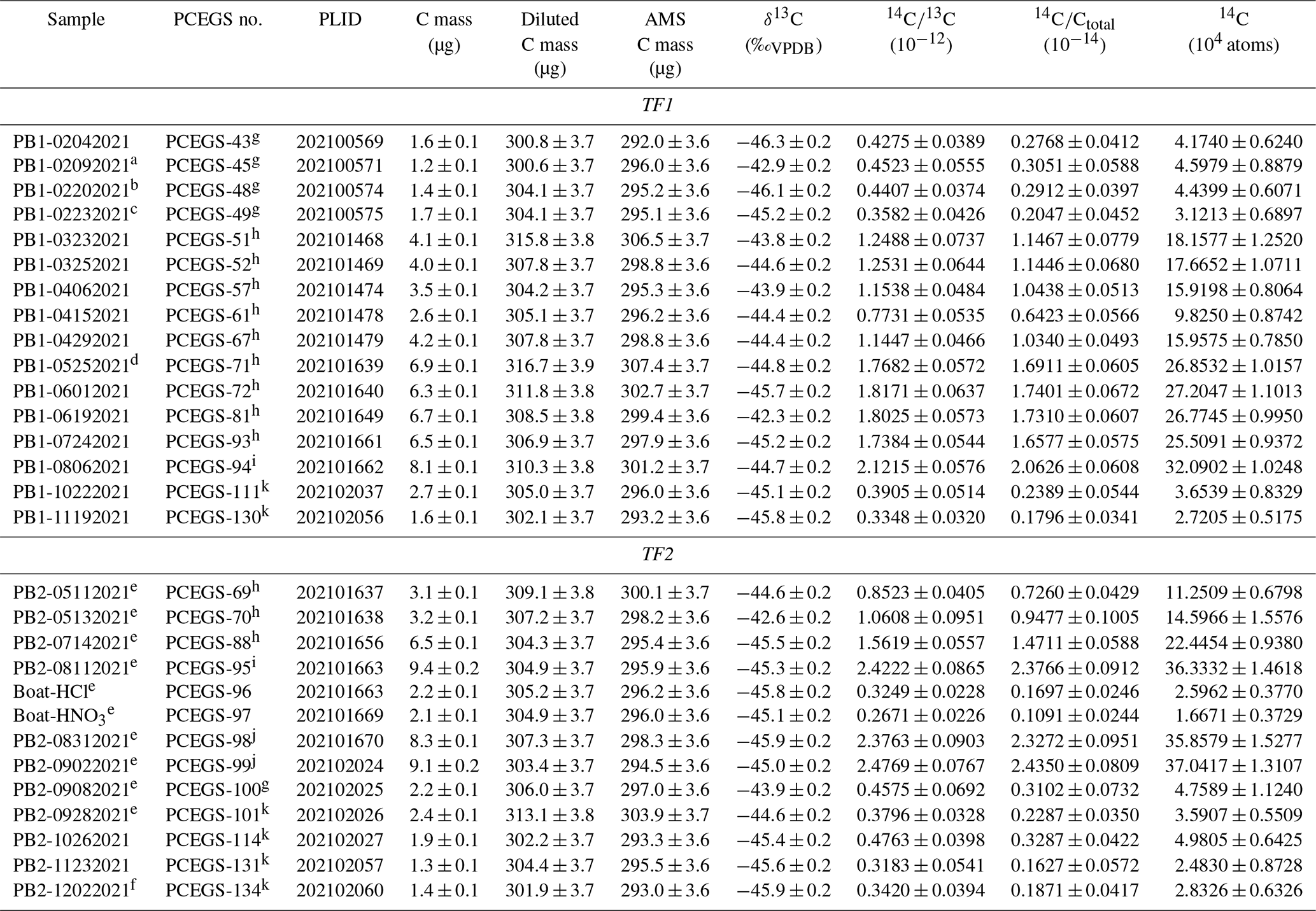
a 1000 ∘C extraction, 3 h. b 1000 ∘C extraction, 4.5 h. c 600 ∘C combustion, 1 h. d 1100 ∘C extraction, 4.5 h. e 1120 ∘C extraction due to furnace miscalibration at 1100 ∘C set point. f 1050 ∘C extraction. g Claisse C-0611-00, Batch C-10001 – “Batch 1” ultrapure. h Claisse C-0610-00, Batch C-17000-10 – “Batch 2” pure. i Claisse C-0610-00, Batch C-19000-10 – “Batch 3” pure. j Claisse C-0611-00, Batch C-19001-10 – “Batch 4” ultrapure. k SPEX Certi-Prep FFB-0000-03, Lot 240920D-2904 ultrapure.
However, subsequent blanks with the new bottle increased in both CO2 yield (ca. 1.5 µg to ca. 4 µg C equivalent) and 14C content (ca. (1.51 ± 0.31) × 105 14C atoms) (Table 4, Fig. 4). Although these values were higher than the initial measurements, they were reproducible on both TF1 and TF2, and thus we continued with normal system operation. Subsequently, the CO2 yields and 14C content inexplicably jumped again to new “stable” values of ca. 6.6 µg C equivalent and (2.66 ± 0.07) × 105 14C atoms, respectively, using TF1, with a similar but slightly lower result with TF2. At that point we tested a second bottle of pure-grade LiBO2 (Claisse C-0610-00, Batch C-19000-10 – “Batch 3” – purchased at the same time as Batch C-17000-10) on procedural blanks in TF1 (PCEGS-94) and TF2 (PCEGS-95), with even higher results of (3.21 ± 0.10) × 105 and (3.63 ± 0.15) × 105 14C atoms, respectively. The higher blanks from Batch C-19000-10 also exhibited higher CO2 yields (ca. 8–9.5 µg C equivalent). In fact, the CO2 yields from each extraction module tracked the 14C atoms quite linearly for all of these experiments, with similar regression fits to each (R2 values of 0.955 and 0.970 for TF1 and TF2, respectively; see Fig. 5).
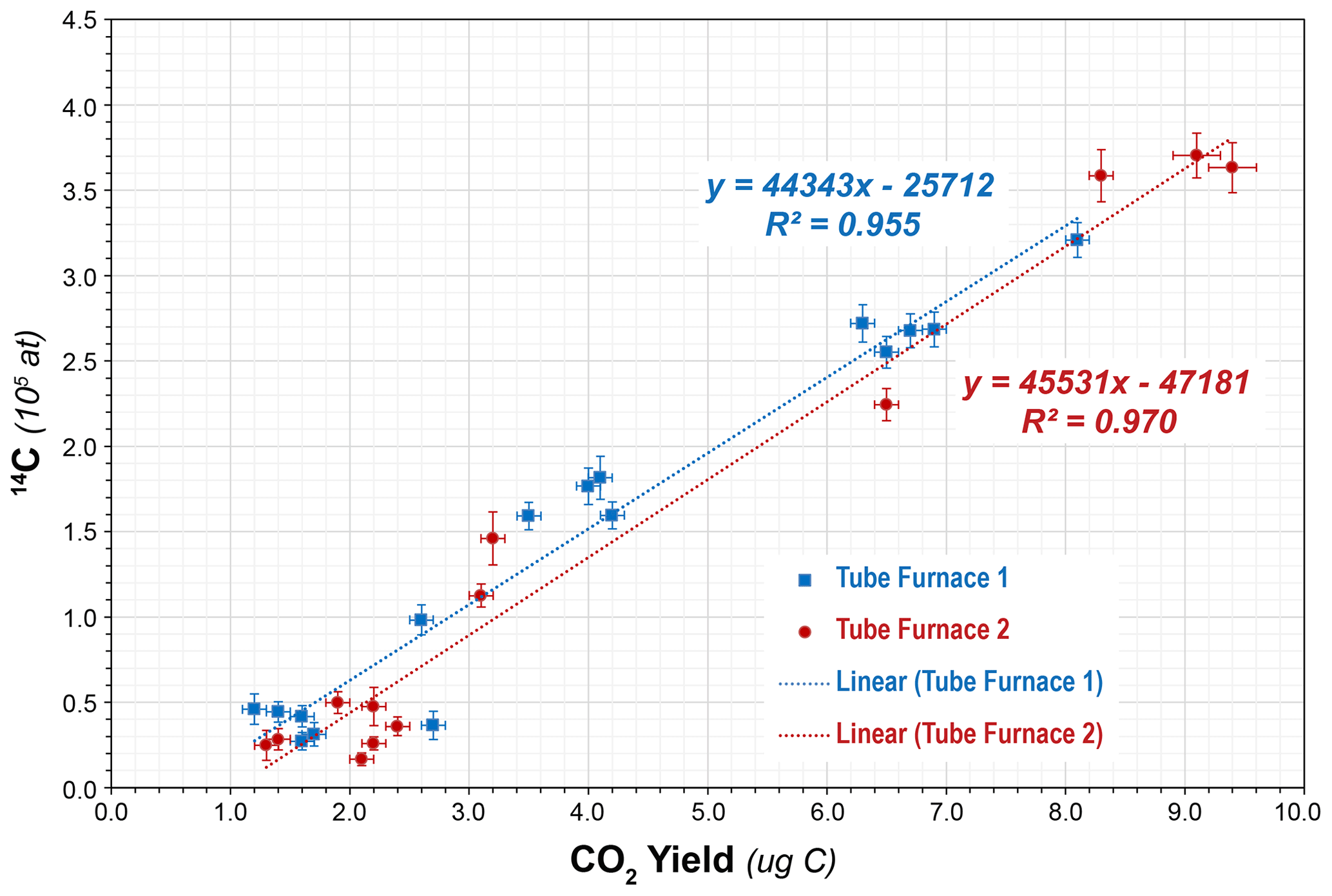
Figure 5Linear fit between CO2 yield (in µg C) vs. procedural blank (in 14C atoms) for tube furnaces 1 and 2 (1σ uncertainties shown).
At that point we paused normal system operations and conducted more basic experiments to try to isolate the source of the increased blanks – was it in the system overall or the LiBO2? Two procedural blanks with everything except for the LiBO2 (boat and sleeve only) – one boat cleaned in 10 % v : v HCl and the other in 10 % v : v HNO3 – both yielded ca. 2.0×104 14C atoms. This indicated that the LiBO2 was the source of the high blank, although the nature of that source and why the blank increased with time is unclear. This is particularly puzzling since the low blanks with boats (compared to the boat-and-sleeve-only blanks) indicate that the degassing step on day 1 should be effective at minimizing atmospheric CO2 contamination from the flux.
We then obtained a new bottle of ultrapure-grade LiBO2 (Claisse C-0611-00, Batch C-19001-10 – “Batch 4”); two blanks from that bottle from TF2 (PCEGS-98 and 99) yielded values comparable to PCEGS-95 – ca. 3.6–3.7 × 105 14C atoms and ca. 8–9 µg C-equivalent yields (Fig. 4, Table 4). Finally, we tried a blank with the remainder of the original bottle of ultrapure-grade LiBO2 (Claisse C-0611-00, Batch C-10001 – “Batch 1”). This experiment (PCEGS-100) exhibited CO2 yield and 14C content comparable to our original tests: 2.2 µg C equivalent and 4.76 ± 1.12 × 104 14C atoms. In consultation with Claisse technical support, we were unable to identify any chemical change in their product or manufacturing process that could have led to the progressively increasing blanks. As such, we identified another vendor, SPEX CertiPrep. We purchased a similar pre-fused ultrapure-grade LiBO2 from them (FFB-0000-03, Lot 240920D-2904) and ran a blank on each extraction module. CO2 yields were comparable to those of the original Claisse ultrapure batch, and 14C contents were slightly improved over that material: ca. 2.5 µg C equivalent and ca. 3.6×104 14C atoms (Table 4, Fig. 4). Subsequent blanks with the new SPEX LiBO2 were generally comparable to or better than those initial measurements, ranging from ca. 2.4×104 to 5.0×104 14C atoms (mean: (3.38 ± 0.92) × 104 14C atoms), and similar to recently published blank measurements from other in situ 14C laboratories using Pt sample boats (e.g., Lupker et al., 2019; Goehring et al., 2019) (Table 4, Fig. 4). Regardless of the ultimate cause of the unexplained blank behavior with the more recent bottles of Claisse LiBO2, we are proceeding with the SPEX ultrapure LiBO2 as our preferred flux.
Late in this process we also discovered that the temperature controller for TF2 was miscalibrated at high-temperature set points, reading 1120 ∘C on an independent Type S thermocouple probe when set to 1100 ∘C. Independent measurement of the lower temperatures for the combustion steps in TF2 agreed with the set points – only the extraction temperatures exhibited the offset. We subsequently adjusted the set point temperatures for extractions to achieve the desired temperature on that furnace (1080 ∘C set point for 1100 ∘C actual and 1035 ∘C set point for 1050 ∘C actual). No such problem was observed with TF1. Results from both blanks and intercomparison materials (Sect. 3.3 below) do not appear to indicate any significant effect from the 20 ∘C excess temperature in the affected TF2 experiments (Figs. 4 and 6, Tables 4 and 5).
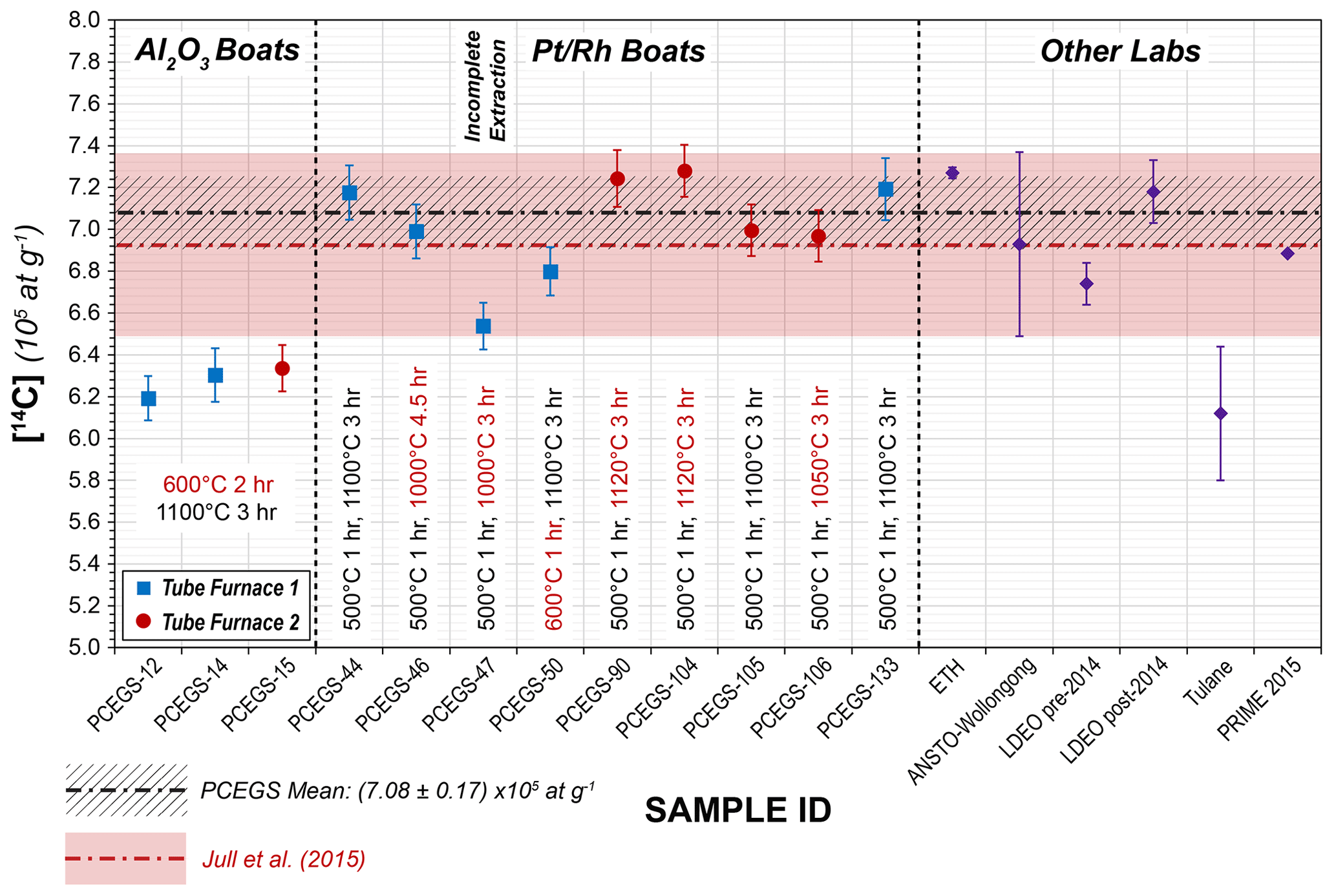
Figure 6CRONUS-A results with experimental details (1σ uncertainties) from this study, with mean values from other studies for comparison. The PCEGS mean value includes all samples except PCEGS-47. Other laboratories' results are listed as follows: ETH (Lupker et al., 2019), ANSTO-Wollongong (Fülöp et al., 2019), LDEO (Lamp et al., 2019), Tulane (Goehring et al., 2019), and PRIME 2015 (Lifton et al., 2015).
Table 5Intercomparison samples. All analyses used 1 h combustion at 500 ∘C and 3 h extraction at 1100 ∘C (unless otherwise noted).
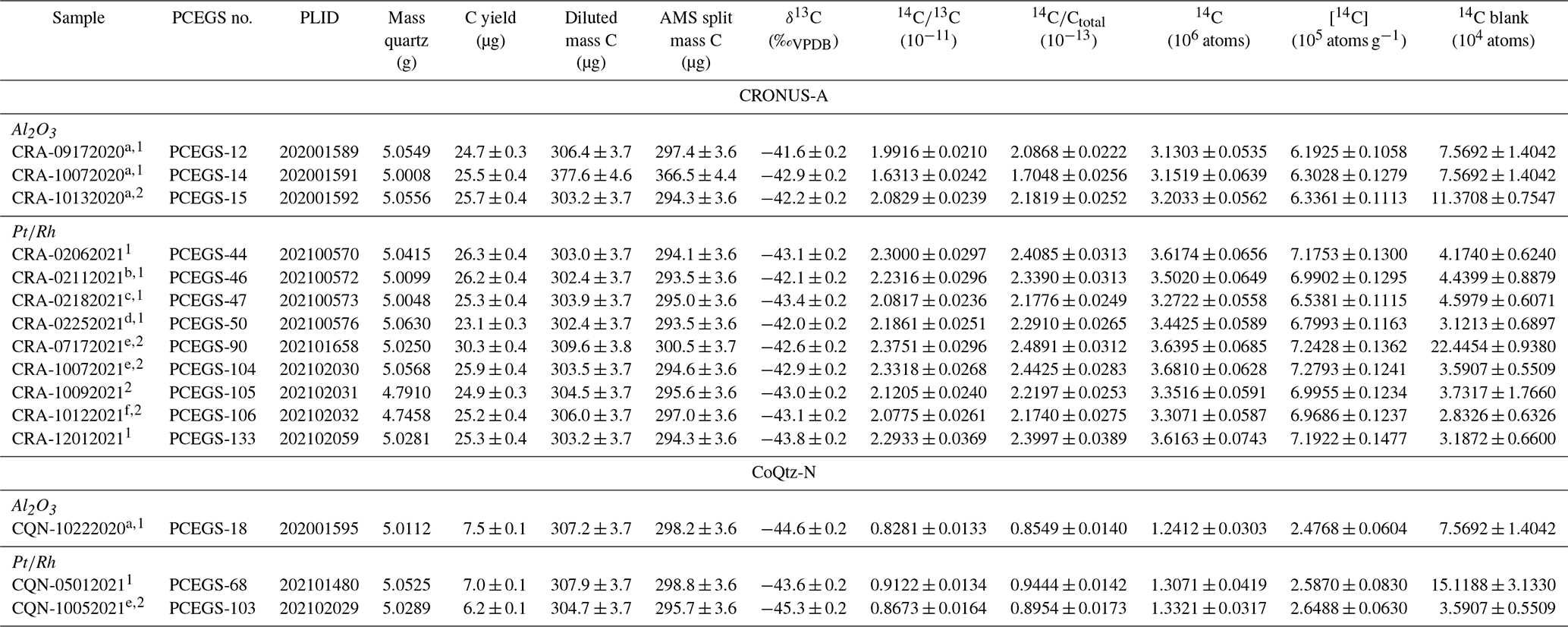
a 2 h at 600 ∘C combustion. b 4.5 h at 1000 ∘C extraction. c 3 h at 1000 ∘C extraction. d 1 h at 600 ∘C combustion. e 3 h at 1120 ∘C extraction. f 3 h at 1050 ∘C extraction. 1 TF1. 2 TF2.
3.3 Extraction experiments with intercomparison materials
While we worked to isolate and understand the source(s) of the time-dependent procedural blanks on our new system, we also set out to better understand the effects of different combustion temperatures and durations on the amount of 14C extracted from the well-studied CRONUS-A intercomparison material derived from Antarctic sandstone bedrock of long exposure duration (Jull et al., 2015). In addition, since the more uniform heating of the boats rendered the LiBO2 flux more broadly aggressive toward the fused-silica sleeves at 1100 ∘C, we tested whether it would be possible to lower the extraction temperature and still achieve full 14C recovery from CRONUS-A. We also initiated measurements at PRIME Lab of the in situ 14C content of the CoQtz-N intercomparison material (e.g., Binnie et al., 2019) using both types of boats. CoQtz-N is derived from a boulder of vein quartz in Namibia, which is again of long exposure duration (Binnie et al., 2019).
3.3.1 CRONUS-A – Al2O3 boats
Initial experiments with the Al2O3 boats used CRONUS-A to test whether the more aggressive combustion procedure described in Sect. 3.2.1 (2 h at 600 ∘C) followed by a 3 h fusion at 1100 ∘C might affect the measured in situ 14C concentrations significantly. Results from both TF1 and TF2 yielded 14C concentrations on the order of 10 % below the consensus value for the material and outside the uncertainty band (Table 5, Fig. 6), suggesting diffusive loss of in situ 14C during the more aggressive low-temperature combustion step. We thus subsequently abandoned that more aggressive procedure in favor of the original 1 h at 500 ∘C combustion step of Lifton et al. (2001) (also Sect. 3.2.2); results from Lifton et al. (2015) using Al2O3 boats with this original procedure are shown for comparison in Fig. 6.
3.3.2 CRONUS-A – Pt Rh boats
Our efforts with the boats largely focused on optimizing extraction temperature and time, again using CRONUS-A as a benchmark (Table 5, Fig. 6). We varied combustion and extraction temperatures and durations using corresponding background corrections appropriate for the procedures used and allowing for the observed procedural blank time dependence.
The experiments with extractions for 3 h at 1100 ∘C and 1120 ∘C and 4.5 h at 1000 ∘C (PCEGS-44, 46, 50, 90, 104, 105, 106, 133; see Table 5, Fig. 6) yielded a mean and standard deviation of (7.08 ± 0.17) × 105 14C atoms g−1 (1σ). An additional extraction test for 3 h at 1000 ∘C (PCEGS-47) yielded a 14C concentration about 8 % lower than this mean but still within the nominal range of results in Jull et al. (2015). However, we judge this extraction as likely to be incomplete as it is outside of the 2σ uncertainty in our mean CRONUS-A analyses, and as such do not consider this further. Another test with a combustion step of 1 h at 600 ∘C and a normal 1100 ∘C extraction (PCEGS-50) yielded a result ca. 4 % below the mean above using a 500 ∘C combustion temperature but within 2σ of that mean and still well within the Jull et al. (2015) range. Excluding PCEGS-50 from the mean above does not significantly change the mean nor these conclusions. After discovering the furnace controller miscalibration for TF2, we also tested CRONUS-A results for TF2 at 1100 ∘C (PCEGS-105) and found them indistinguishable from the mean. Finally, given the less aggressive corrosion of the quartz sleeve from tests at 1050 ∘C (Fig. 3b), we also tested CRONUS-A extraction for 3 h at 1050 ∘C (PCEGS-106), with results being indistinguishable from our overall mean (Table 5, Fig. 6). We thus have switched to a 3 h at 1050 ∘C extraction temperature and duration going forward.
Our CRONUS-A results are consistent with the consensus value and range of Jull et al. (2015), i.e., (6.93 ± 0.44) × 105 14C atoms g−1, as well as with the mean of our previous results at PRIME Lab (Lifton et al., 2015), i.e., (6.89 ± 0.04) × 105 14C atoms g−1. In addition, these new results are consistent with recent measurements by Lupker et al. (2019), Fülöp et al. (2019), and Lamp et al. (2019) (Fig. 6). Like those other studies, they also disagree with the CRONUS-A measurements of Goehring et al. (2019), for reasons yet to be determined (Fig. 6).
3.3.3 CoQtz-N
Our three results for the CoQtz-N intercomparison material spanned the period discussed in this work (Table 5). An initial analysis using an Al2O3 boat and the more aggressive 2 h combustion at 600 ∘C returned a lower concentration (2.48 ± 0.06) × 105 14C atoms g−1 than the two experiments at 500 ∘C/1100 ∘C (TF1) and 500 ∘C/1120 ∘C (TF2), which agree within 1σ measurement uncertainties and yield a mean value of (2.62 ± 0.04) × 105 14C atoms g−1. Interestingly, the Al2O3 result with the more aggressive combustion step is only about 5 % lower than the mean CoQtz-N result (uncertainties overlap at 2σ), while the Al2O3 analyses of CRONUS-A are 9 % lower than the corresponding nominal value. The source of this difference is not clear, but it likely reflects intrinsic differences in diffusive properties of the quartz from each sample.
We only found one other study in which in situ 14C had been measured in CoQtz-N (Schiffer et al., 2020), but that study provides incomplete experimental details and only a plot of concentrations vs. quartz mass without any tabulated data. The four measured values presented for 1 g of CoQtz-N appear to span concentrations ca. 3×105 to over 4×105 14C atoms g−1 – well above our measured values. The source of this discrepancy merits further investigation but is currently difficult to evaluate without complete experimental details.
This study details key characteristics of and procedures in use for the new in situ 14C extraction system at PRIME Lab (PCEGS), and presents results of initial testing of procedural blanks and intercomparison materials. We compare results using the original single-use Al2O3 sample boats employed since Lifton et al. (2001) with those from a new set of reusable 90 %Pt 10 %Rh alloy sample boats.
It is clear from these experiments that the reusable boats provide distinct advantages over the Al2O3 boats, supporting the suggestions of Goehring et al. (2019). First, the boats appear to heat much more aggressively than the sintered Al2O3 ceramic boats, likely leading to more uniform heating of the contents. The boats also appear to reduce or eliminate a significant component of the blank variability associated with the sintered ceramics, perhaps associated with small amounts of atmospheric carbon potentially incorporated into the ceramics during manufacture. Taken together, the aggressive uniform heating and purity of the alloy allow for improved analytical reproducibility, allowing robust identification of systematic influences on background signals that we were previously unable to resolve with the Al2O3 boats.
Using the boats, we demonstrated that time-dependent increases in procedural blanks were tied directly to specific batches of LiBO2 fluxes manufactured by Claisse. The time dependence did not appear to reflect flux purity, but instead some presently unknown characteristic of the Claisse fluxes appears to have changed since the original batch we used for our early experiments. Subsequent analyses with LiBO2 from an alternate supplier, SPEX CertiPrep, yielded consistently low procedural blanks on the order of (3.4 ± 0.9) × 104 14C atoms, and we have switched to that flux going forward.
We also analyzed two intercomparison materials as part of our initial experiments to confirm compatibility with earlier results from this lab and from others. Using both Al2O3 and boats, we focused mainly on CRONUS-A but also made initial measurements for our laboratory of the newer CoQtz-N intercomparison material. We first tested CRONUS-A in Al2O3 boats using a more aggressive combustion procedure than typically used (2 h at 600 ∘C vs. 1 h at 500 ∘C) and found significantly lower 14C concentrations from the high-temperature extraction relative to the nominal value of Jull et al. (2015), likely due to diffusive loss during the more aggressive low-temperature step. Abandoning that aggressive procedure in favor of the shorter 500 ∘C combustion and switching to the boats, we then explored various time–temperature combinations for the high-temperature extraction step with CRONUS-A. Results for 3 h extractions at temperatures ranging from 1050 to 1120 ∘C and 4.5 h at 1000 ∘C yielded similar results, in agreement with the consensus value and with published results from most laboratories, including those using our previous extraction system (Lifton et al., 2015). On the other hand, an extraction for 3 h at 1000 ∘C yielded a significantly lower concentration than the other analyses in this study, suggesting incomplete extraction for those conditions. Based on these results, our preferred technique is now combustion for 1 h at 500 ∘C followed by a 3 h extraction at 1050 ∘C.
The initial analysis of CoQtz-N at PRIME Lab used the more aggressive combustion step but displayed less diffusive loss (relative to our analyses with boats) than CRONUS-A did with that procedure, suggesting variable low-temperature diffusion behavior among samples. Subsequently, internally consistent results were achieved with CoQtz-N using boats, with approximately 60 % lower 14C concentrations than CRONUS-A. However, additional analyses of this material from this and other labs are clearly needed to work toward a consensus value.
All data are presented in Tables 2–5 within this paper.
This study was conceived by NL and JW. Sample preparation and analysis were done by NL and AK. NL analyzed the data and wrote the manuscript, with contributions from AK and JW.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors gratefully acknowledge advice and technical assistance from Marc Caffee and Tom Woodruff of PRIME Lab.
Funding for this research was provided by US National Science Foundation grant EAR-1560658 and a Purdue University Laboratory and University Core Facility Research Equipment Program grant (2017-2018).
This paper was edited by Hella Wittmann-Oelze and reviewed by Jennifer Lamp and Irene Schimmelpfennig.
Binnie, S. A., Dewald, A., Heinze, S., Voronina, E., Hein, A., Wittmann, H., Blanckenburg, F. von, Hetzel, R., Christl, M., Schaller, M., Léanni, L., Team, A., Hippe, K., Vockenhuber, C., Ivy-Ochs, S., Maden, C., Fülöp, R.-H., Fink, D., Wilcken, K. M., Fujioka, T., Fabel, D., Freeman, S. P. H. T., Xu, S., Fifield, L. K., Akçar, N., Spiegel, C., and Dunai, T. J.: Preliminary results of CoQtz-N: A quartz reference material for terrestrial in-situ cosmogenic 10Be and 26Al measurements, Nucl. Instrum. Meth. B, 456, 203–212, https://doi.org/10.1016/j.nimb.2019.04.073, 2019.
Donahue, D. J., Jull, A. J. T., and Toolin, L.: Radiocarbon measurements at the University of Arizona AMS facility, Nucl. Instrum. Meth. B, 52, 224–228, 1990.
Donahue, D. J., Beck, J. W., Biddulph, D., Burr, G. S., Courtney, C., Damon, P. E., Hatheway, A., Hewitt, L., Jull, A. J. T., Lange, T., Lifton, N., Maddock, R., McHargue, L., O'Malley, J. M., and Toolin, L.: Status of the NSF-Arizona AMS laboratory, Nucl. Instrum. Meth. B, 123, 51–56, 1997.
Fülöp, R., Naysmith, P., Cook, G., and Fabel, D.: Update on the performance of the SUERC in situ cosmogenic 14C extraction line, Radiocarbon, 52, 1288–1294, 2010.
Fülöp, R. H., Wacker, L., and Dunai, T. J.: Progress report on a novel in situ 14C extraction scheme at the University of Cologne, Nucl. Instrum. Meth. B, 361, 20–24, https://doi.org/10.1016/j.nimb.2015.02.023, 2015.
Fülöp, R.-H., Fink, D., Yang, B., Codilean, A. T., Smith, A., Wacker, L., Levchenko, V., and Dunai, T. J.: The ANSTO – University of Wollongong in-situ 14C extraction laboratory, Nucl. Instrum. Meth. B, 438, 207–213, https://doi.org/10.1016/j.nimb.2018.04.018, 2019.
Goehring, B. M., Wilson, J., and Nichols, K.: A fully automated system for the extraction of in situ cosmogenic carbon-14 in the Tulane University cosmogenic nuclide laboratory, Nucl. Instrum. Meth. B, 455, 284–292, https://doi.org/10.1016/j.nimb.2019.02.006, 2019.
Hippe, K. and Lifton, N. A.: Calculating Isotope Ratios and Nuclide Concentrations for In Situ Cosmogenic 14C Analyses, Radiocarbon, 56, 1167–1174, https://doi.org/10.2458/56.17917, 2014.
Hippe, K., Kober, F., Baur, H., Ruff, M., Wacker, L., and Wieler, R.: The current performance of the in situ 14C extraction line at ETH, Quat. Geochronol., 4, 493–500, https://doi.org/10.1016/j.quageo.2009.06.001, 2009.
Hippe, K., Kober, F., Wacker, L., Fahrni, S. M., Ivy-Ochs, S., Akcar, N., Schluchter, C., and Wieler, R.: An update on in situ cosmogenic 14C analysis at ETH Zürich, Nucl. Instrum. Meth. B, 294, 81–86, 2013.
Jull, A. J. T., Scott, E. M., and Bierman, P.: The CRONUS-Earth inter-comparison for cosmogenic isotope analysis, Quat. Geochronol., 26, 3–10, https://doi.org/10.1016/j.quageo.2013.09.003, 2015.
Lamp, J. L., Young, N. E., Koffman, T., Schimmelpfennig, I., Tuna, T., Bard, E., and Schaefer, J. M.: Update on the cosmogenic in situ 14C laboratory at the Lamont-Doherty Earth Observatory, Nucl. Instrum. Meth. B, 456, 157–162, https://doi.org/10.1016/j.nimb.2019.05.064, 2019.
Lifton, N., Jull, A., and Quade, J.: A new extraction technique and production rate estimate for in situ cosmogenic 14C in quartz, Geochim. Cosmochim. Ac., 65, 1953–1969, 2001.
Lifton, N., Goehring, B., Wilson, J., Kubley, T., and Caffee, M.: Progress in automated extraction and purification of in situ 14C from quartz: Results from the Purdue in situ 14C laboratory, Nucl. Instrum. Meth. B, 361, 381–386, https://doi.org/10.1016/j.nimb.2015.03.028, 2015.
Lupker, M., Hippe, K., Wacker, L., Steinemann, O., Tikhomirov, D., Maden, C., Haghipour, N., and Synal, H.-A.: In-situ cosmogenic 14C analysis at ETH Zürich: Characterization and performance of a new extraction system, Nucl. Instrum. Meth. B, 457, 30–36, https://doi.org/10.1016/j.nimb.2019.07.028, 2019.
Nichols, K. A. and Goehring, B. M.: Isolation of quartz for cosmogenic in situ 14C analysis, Geochronology, 1, 43–52, https://doi.org/10.5194/gchron-1-43-2019, 2019.
Pigati, J., Lifton, N., Jull, A. J. T., and Quade, J.: A simplified in situ cosmogenic 14C extraction system, Radiocarbon, 52, 1236–1243, 2010.
Santos, G. M., Southon, J. R., Druffel-Rodriguez, K., Griffin, S., and Mazon, M.: Magnesium Perchlorate as an Alternative Water Trap in AMS Graphite Sample Preparation: A Report on Sample Preparation at KCCAMS at the University of California, Irvine, Radiocarbon, 46, 165–174, 2004.
Schiffer, M., Stolz, A., López, D. A., Spanier, R., Herb, S., Müller-Gatermann, C., Heinze, S., Binnie, S., Melchert, J., Kivel, N., Schumann, D., Rethemeyer, J., Dunai, T., and Dewald, A.: Method developments for accelerator mass spectrometry at CologneAMS, burial dating and ultra-small 14CO2 samples, Glob. Planet. Change, 184, 103053, https://doi.org/10.1016/j.gloplacha.2019.103053, 2020.
Southon, J.: Graphite reactor memory – Where is it from and how to minimize it?, Nucl. Instrum. Meth. B, 259, 288–292, 2007.