the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Aug 2024
| 12 Aug 2024
Technical note: Optimizing the in situ cosmogenic 36Cl extraction and measurement workflow for geologic applications
Joseph M. Licciardi
Alan J. Hidy
Tyler S. Anderson
In situ cosmogenic 36Cl analysis by accelerator mass spectrometry (AMS) is routinely employed to date Quaternary surfaces and assess rates of landscape evolution. However, standard laboratory preparation procedures for 36Cl dating require the addition of large amounts of isotopically enriched chlorine spike solution; these solutions are expensive and increasingly difficult to acquire from commercial sources. In addition, the typical workflow for 36Cl dating involves measuring both and concurrently on the high-energy (post-accelerator) end of the AMS system, but determinations using this technique can be complicated by isotope fractionation and system memory during measurement. The traditional workflow also does not provide 36Cl extraction laboratories with the data needed to calculate native Cl concentrations in advance of measurements. In light of these concerns, we present an improved workflow for extracting and measuring chlorine in geologic materials. Our initial step is to characterize on sample aliquots of up to ∼1 g prepared in Ag(Cl, Br) matrices, which greatly reduces the amount of isotopically enriched spike solution required to measure native Cl content in each sample. To avoid potential issues with isotope fractionation through the accelerator, is measured on the low-energy, pre-accelerator end of the AMS line. Then, for measurements, we extract Cl as AgCl or Ag(Cl, Br) in analytical batches with a consistent total Cl load across all samples; this step is intended to minimize source memory effects during measurements and allows the preparation of AMS standards that are customized to match known Cl contents in the samples. To assess the efficacy of this extraction and measurement workflow, we compare chlorine isotope ratio measurements on seven geologic samples prepared using standard procedures and the updated workflow. Measurements of and are consistent between the two workflows, and values measured using our methods have considerably higher precision than those measured following standard protocols. The chemical preparation and measurement workflow presented here (1) reduces the amount of isotopically enriched chlorine spike used per rock sample by up to 95 %; (2) identifies rocks with high native Cl concentrations, which may be lower priority for 36Cl surface exposure dating, at an early stage of analysis; and (3) allows laboratory users to maintain control over the total chlorine content within and across analytical batches. These methods can be incorporated into existing laboratory and AMS protocols for 36Cl analyses and will increase the accessibility of 36Cl dating for geologic applications.
- Article
(2839 KB) - Full-text XML
- BibTeX
- EndNote





Cosmogenic 36Cl is widely used within the geosciences to determine the duration of the surface exposure of Quaternary features such as glacial deposits (Barth et al., 2019; Phillips et al., 1997; Small et al., 2016), lava flows (Parmelee et al., 2015; Singer et al., 2018), landslides (Ivy-Ochs et al., 2009; Zerathe et al., 2014; Pánek et al., 2018), terraces (Kozaci et al., 2007; Robertson et al., 2019), and fault scarps (Benedetti et al., 2002; Mitchell et al., 2001; Schlagenhauf et al., 2011). Over the past few decades, 36Cl has also emerged as the primary isotope for constraining rates of landscape evolution in carbonate settings (Ben-Asher et al., 2021; Marrero et al., 2018; Stone et al., 1996). 36Cl offers several advantages over other commonly used cosmogenic isotopes (e.g., 10Be) for surficial geochronology. 36Cl is produced by multiple reactions in a wide variety of minerals, including orthoclase, plagioclase, and calcite (Gosse and Phillips, 2001), enabling its use in dating mineral separates and whole-rock samples of nearly any lithology. The high production rate of 36Cl (Marrero et al., 2016) also allows age determinations on young materials (e.g., Price et al., 2022). Additionally, accelerator mass spectrometry (AMS) measurements of chlorine (prepared as AgCl) have low detection limits and high beam currents (Finkel et al., 2013). Finally, recent advances in 36Cl production rate calibrations (Marrero et al., 2016) and the availability of web-based calculators (e.g., CRONUScalc, https://cronus.cosmogenicnuclides.rocks/2.1/html/cl/, last access: 26 February 2024; CRONUSEarth, http://stoneage.ice-d.org/math/Cl36/v3/v3_Cl36_age_in.html, last access: 26 February 2024; CREP, https://crep-dev.otelo.univ-lorraine.fr/#/init, last access: 26 February 2024) have enabled surface exposure ages or erosion rates to be determined with relative ease once total sample Cl, 36Cl concentration, and elemental concentrations are obtained.
In situ 36Cl concentrations are typically measured via AMS methods on targets prepared in an AgCl matrix (Fig. 1; Licciardi et al., 2008). To ensure that Cl isotope ratios are well above laboratory blank values, consistent sample masses are prepared for Cl isotope analysis; depending on anticipated 36Cl inventories (which are a function of exposure duration, altitude, and sample composition), each sample usually consists of ∼10–20 g of milled rock for whole-rock silicates or ∼5–10 g of isolated mineral separates. Rock samples are spiked with isotopically enriched Cl carrier solution such that total sample Cl (from ) and 36Cl concentrations (from or ) can be determined through isotope dilution methods (Faure and Mensing, 2005). At the University of New Hampshire and the Center for AMS (CAMS) at Lawrence Livermore National Laboratory (LLNL), geologic samples have historically been spiked with ∼750–1000 µg of Cl from a 37Cl-enriched solution with a of approximately 1, which is substantially lower than the natural of 3.127. Following standard protocols (Licciardi et al., 2008), Cl is extracted from the samples as AgCl. After shipment to CAMS, the AgCl precipitates are packed into AgBr plugs that are pressed into open-faced stainless steel cathodes. Both and are then measured simultaneously on the high-energy (i.e., post-accelerator) end of the 10 MeV Tandem Van de Graaff accelerator at CAMS, and isotope extraction laboratories receive and results after all measurements are completed.
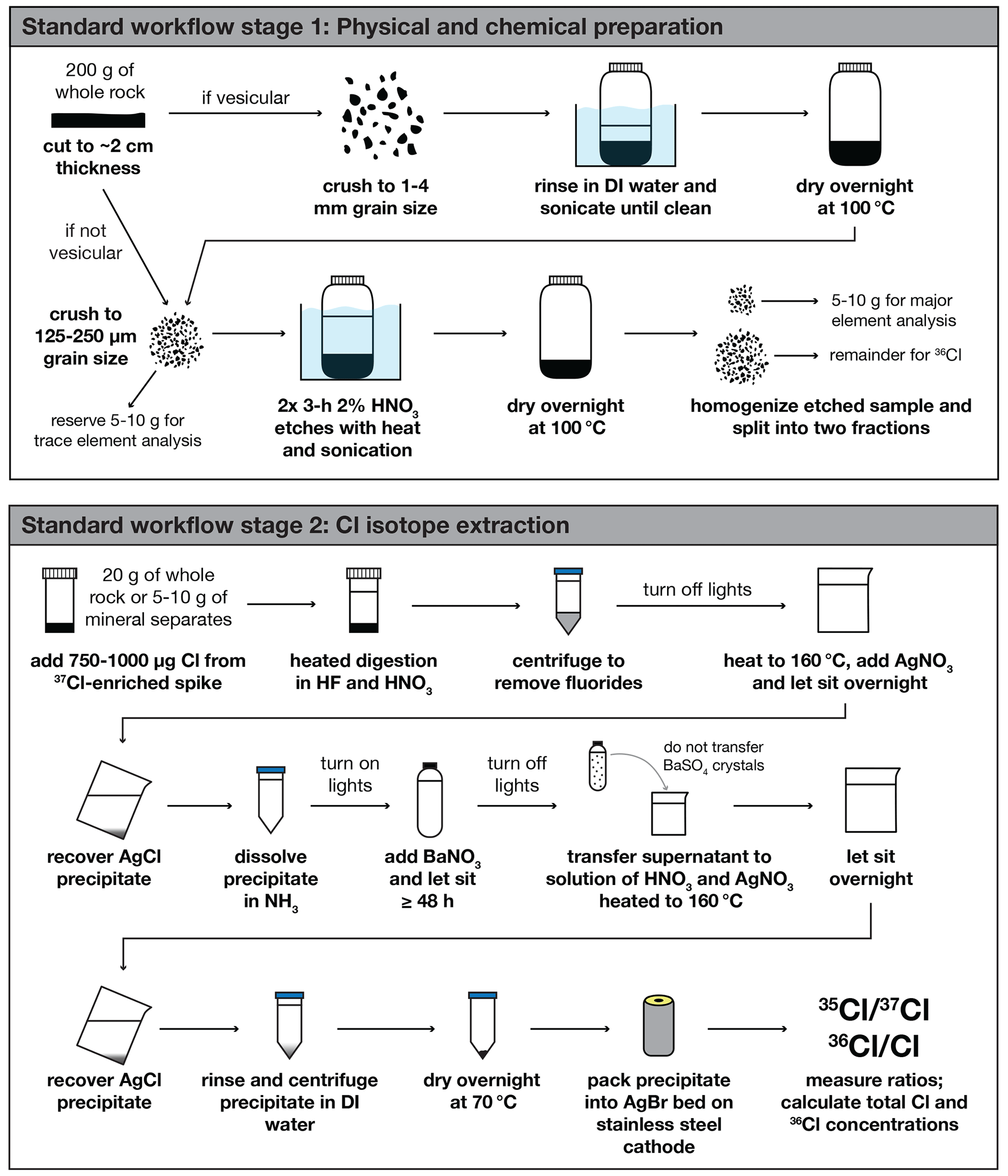
Figure 1Schematic of the standard workflow for cosmogenic 36Cl analysis based on Licciardi et al. (2008). Black arrows indicate the order of steps for each stage of the process. Note that while we use a 37Cl-enriched spike solution for the stable Cl extraction, these procedures are also suitable for use with a 35Cl-enriched spike.
While this workflow has proven useful over many years of Cl measurements at CAMS, there are several areas in which procedural and performance improvements are possible. Firstly, few commercial sources of 37Cl-enriched solutions exist with negligible 36Cl concentrations that are acceptable as carriers for 36Cl sample preparation, and available source materials are extremely expensive. Alternatively, commercial sources of 35Cl-enriched carriers are more readily obtained and available with negligible 36Cl concentrations. However, when 36Cl, 35Cl, and 37Cl are measured simultaneously at the high-energy end of an AMS system, it is undesirable to use carrier enriched in the more abundant isotope (35Cl). This is because it inflates the difference in intensity between 35Cl− and 37Cl− beams injected into the accelerator, leading to increased instability in maintaining terminal voltage. Considering these issues, the supply of 37Cl-enriched spike that is currently being used for sample preparation in extraction laboratories is limited and/or undesirable unless laboratory users have the means to invest substantial sums for new isotopically enriched chlorine solutions. Secondly, in the sample preparation methods outlined above, the native Cl content in each geologic sample is not known in advance of the measurements; hence it is not possible to control total Cl in an analytical batch of AMS targets. Variable total Cl among samples can cause AMS memory effects after the ionization of unexpectedly high Cl targets (Arnold et al., 2010; Finkel et al., 2013), and the reduction of these memory effects involves longer measurement times per sample. Finally, measurements of stable Cl ratios on the high-energy, post-accelerator portion of the AMS can be affected by isotope fractionation in the terminal stripper at high (>40 µA) 35Cl beam currents, leading to inaccurate determinations of (Wilcken et al., 2013). Accurate measurements are essential for determining relative contributions of different 36Cl production pathways to the cosmogenic 36Cl inventory in a sample and for deriving the cosmogenic 36Cl concentration and exposure ages.
In this technical note, we present an improved laboratory and analytical workflow for measuring Cl isotope concentrations in silicate rocks via AMS. This workflow is similar to procedures developed at the University of Washington Cosmogenic Isotope Laboratory (Stone et al., 1996; Stone, 2001) but with a key difference being that we strongly encourage laboratory users to prepare subsamples for measurements in advance of analyses. While this workflow was implemented specifically for silicates, where hydrofluoric acid (HF) is required for full digestion, it is suitable for carbonates as well by simply modifying the digestion step to exclude HF. Through a set of experiments on geologic samples, we demonstrate that the updated workflow provides comparable and, in some cases, more precise results than the standard workflow. A key finding of our experiments is that, compared to standard methods, our workflow reduces the use of costly isotopically enriched Cl spike solution by up to 95 %, which should increase the accessibility of 36Cl dating for geologic applications, as laboratory users can prepare more samples with their existing supplies.
Our improved cosmogenic Cl workflow involves measuring sample chloride concentrations via isotope dilution on a small (∼1 g) aliquot of rock prior to digestion of the full-rock sample (Fig. 2). analyses are performed on the low-energy (pre-accelerator) end of an AMS with as little as 50 µg of isotopically enriched Cl added to the target. This reduction in Cl is compensated by bulking the sample with bromine (from a carrier made with commercially sourced NH4Br) and co-precipitating as Ag(Cl, Br) to facilitate consistent sample handling and fully packed targets. Once total Cl concentrations have been determined for each sample, rock samples are prepared for measurements using a modified version of standard 36Cl methods, with an optional addition of NH4Br carrier at the dissolution stage for samples with low total Cl loads. measurements are made on AgCl or Ag(Cl, Br) targets containing a minimum of 500 µg Cl but ideally 750–1000 µg Cl as AgCl.
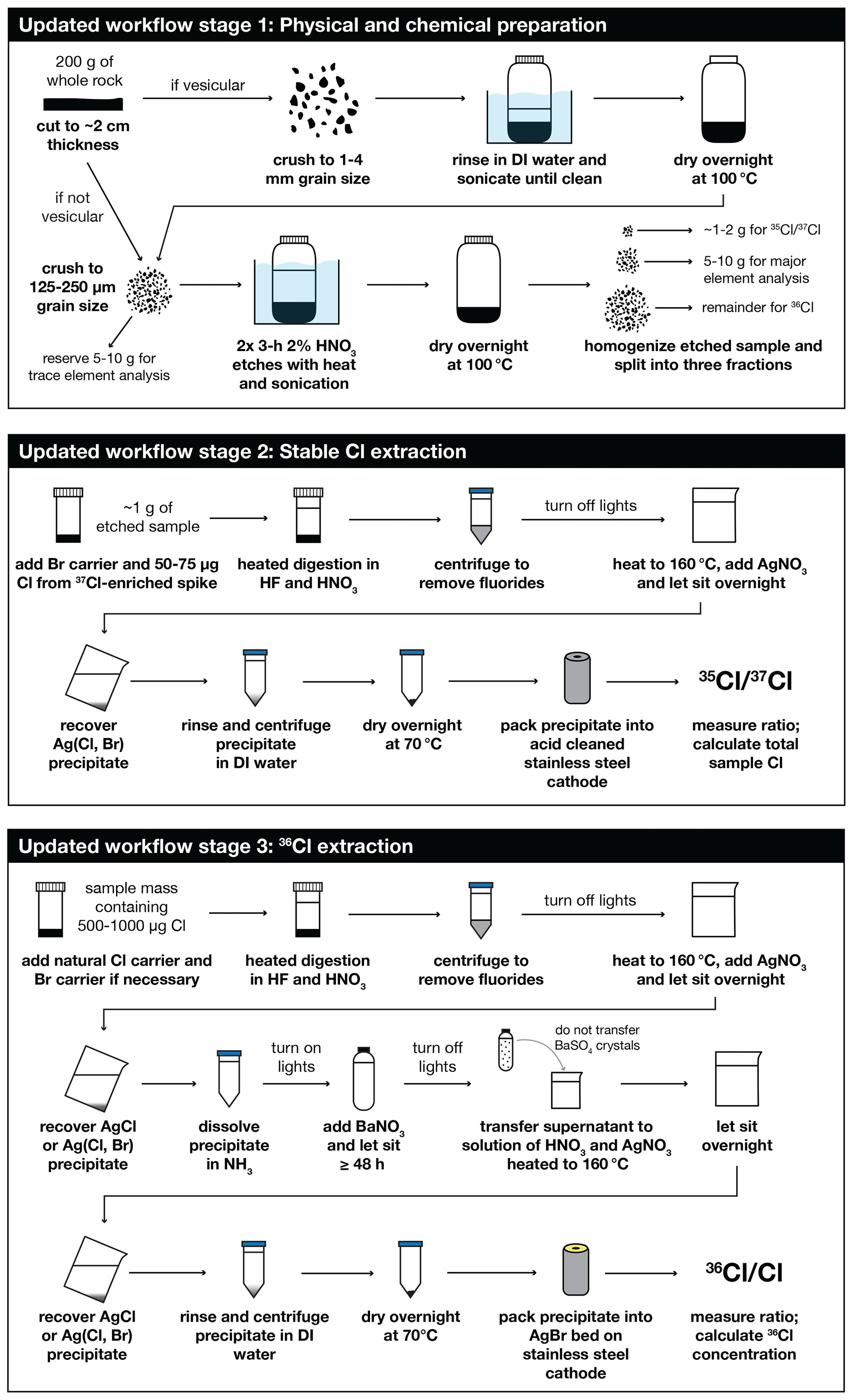
Figure 2Schematic of the updated workflow for cosmogenic 36Cl analysis presented here. Black arrows indicate the order of steps for each stage of the process. Note that while we use a 37Cl-enriched spike solution for the stable Cl extraction, these procedures are also suitable for use with a 35Cl-enriched spike.
This approach offers several advantages compared to the traditional workflow. Because Cl concentrations are determined before preparing a rock sample for 36Cl analysis, there is no need to add an isotopically enriched carrier solution to the full sample. Instead, if additional Cl is required, a carrier solution containing natural-ratio Cl can be used (Stone, 2001). The natural-ratio Cl solution can be made with widely available and inexpensive material such as mined rock salt that is typically low in 36Cl. With this approach, each sample in an analytical measurement batch is tuned to contain a similar amount of total chlorine; the 37Cl-enriched spike solution is conserved; and the presence of significant 36Cl in the enriched spike solution becomes a non-issue, since it is used only for the stable Cl aliquots and not the target measured for .
We tested this workflow on seven whole-rock geologic samples with varying silicate lithologies and a wide range of expected 36Cl inventories. For comparison, we also measured stable Cl and cosmogenic 36Cl on splits of the same samples prepared using standard procedures (Fig. 1; Licciardi et al., 2008). All chlorine isotope ratio measurements were performed at LLNL-CAMS between July 2020 and December 2023. The setup of the CAMS accelerator and the operational parameters for measurements are described in detail in Anderson et al. (2022).
2.1 Cl dilution series
To assess the accuracy of measurements on the low-energy (i.e., pre-accelerator) portion of the AMS, we prepared a dilution series of four samples using varying amounts of 37Cl-enriched spike, a natural-ratio Cl carrier, and a NH4Br carrier. For this dilution series, we used a 37Cl-enriched solution prepared at the University of New Hampshire (“Wildcat spike”). The Wildcat spike has a of 1.001 and a Cl concentration of 942 ± 15 µg g−1 (measured via ICP-OES; n=4; average ± 1 standard deviation). To make the Wildcat spike, we combined 37Cl-enriched NaCl powder with a natural-ratio Cl carrier solution to achieve the desired of 1.001. The isotopically enriched NaCl powder was purchased in October 2004 from Oak Ridge National Laboratory (ORNL) and had an atomic assay of 98.21 % 37Cl and 1.79 % 35Cl (ORNL batch 198501). The natural-ratio Cl carrier solution (“Weeks Island Halite carrier”) was made at the University of New Hampshire by dissolving NaCl obtained from a mine in Weeks Island, Louisiana, in deionized water. The Cl concentration in the Weeks Island Halite carrier is 1436 ± 9 µg g−1 (measured via ICP-OES; n=4; average ± 1 standard deviation). In the dilution series, we adjusted the amounts of the Wildcat spike and Weeks Island Halite carriers such that the total Cl in each sample was ∼150 µg, while the expected values ranged from 1.001 to 2.510 (Table 1). All samples were bulked with ∼4000 µg Br from a 10 000 µg g−1 NH4Br carrier solution.
Table 1Results of measurements of a dilution series to test low-energy AMS performance across a range of values expected in geologic samples.
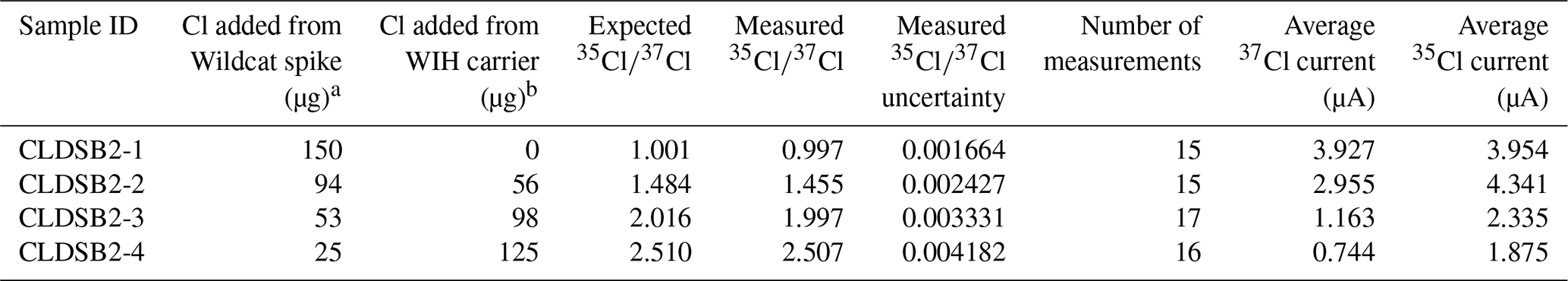
a The “Wildcat spike” has a measured of 1.001 and a Cl concentration of 942 ± 15 µg g−1. b The “Weeks Island Halite” (WIH) carrier has a of 3.127 (i.e., natural ratio) and a Cl concentration of 1436 ± 9 µg g−1.
To quantify the magnitude of Cl contamination in our bromine carrier, we prepared a second dilution series using the 37Cl-enriched Wildcat spike and the NH4Br carrier. We prepared five samples for this dilution series with Cl loads ranging from ∼50 to 150 µg Cl. All samples received ∼0.4 g of 10 000 µg g−1 NH4Br solution (∼4000 µg Br; Table 2). For both dilution series, after the carrier additions, the solutions were acidified in 8 mL of 2 M HNO3. To precipitate Ag(Cl, Br), ∼1 mL of 5 % AgNO3 was added to each solution under low-light conditions and left to precipitate for at least 12 h. The precipitates were then rinsed, dried, and packed into acid-cleaned stainless steel cathodes for measurement at CAMS.
Table 2 measurements of a dilution series to test Cl contamination in the commercial NH4Br source used in the preparation of geologic test samples.
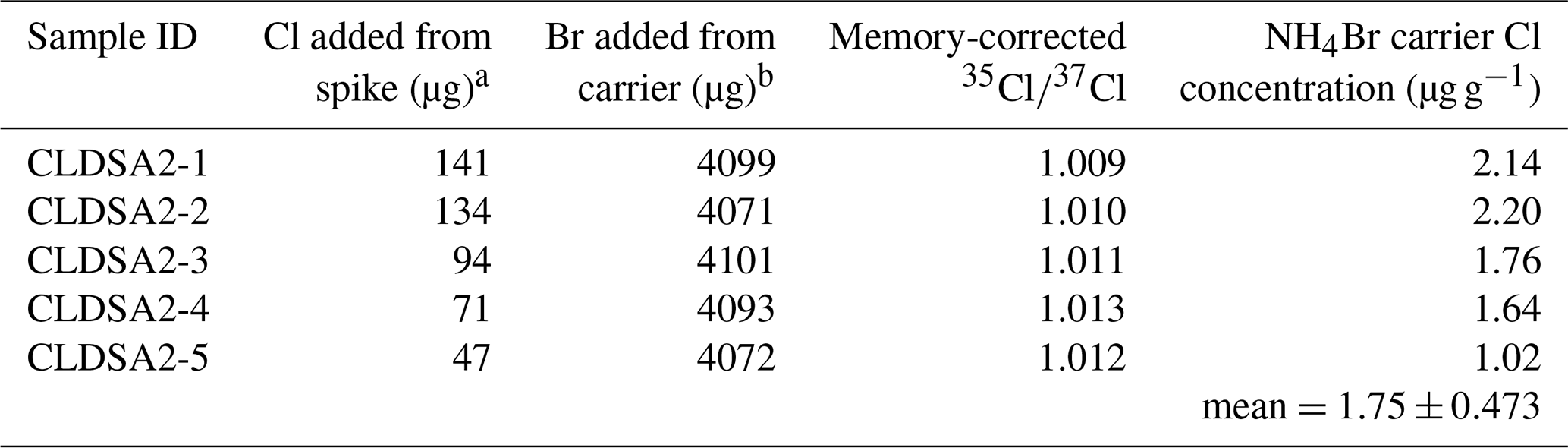
a Samples were spiked with the “Wildcat spike,” which has a measured of 1.001 and a Cl concentration of 942 ± 15 µg g−1. b The carrier solution has a concentration of ∼10 000 µg g−1 NH4Br.
2.2 Preparation of test samples
We prepared a set of seven geologic samples for and 36Cl analyses (Fig. 2). Four samples (19SEAK-01, -02, -12, and -13) were collected in 2019 from two locations in coastal southeastern Alaska. 19SEAK-01 and 19SEAK-02 were sampled from erratic boulders of olivine basalt on Suemez Island (Eberlein et al., 1983) that were deposited by the Cordilleran Ice Sheet during the last deglaciation (Walcott et al., 2022). Samples 19SEAK-12 and 19SEAK-13 were obtained from the surface of a vesicular plagioclase basalt flow that was emplaced in the eastern Mount Edgecumbe volcanic field sometime during the late Pleistocene (Riehle et al., 1989). Three rhyolite samples (YGT18-31, -32, and -33) were collected in 2018 from the Yellowstone Plateau. These samples were taken from the top surfaces of erratic boulders deposited during the most recent deglaciation of the Yellowstone ice cap (Licciardi and Pierce, 2018).
After initial physical and chemical preparation, we split each rock sample into two fractions. We prepared the “A” splits (e.g., 19SEAK-01A) using standard procedures (Fig. 1; Stone et al., 1996; Licciardi et al., 2008), where targets are prepared in an AgCl matrix and stable Cl ratios and cosmogenic 36Cl are measured concurrently on the post-accelerator end of the AMS. We used the “B” splits (e.g., 19SEAK-01B) to test the updated workflow (Fig. 2). For these samples, we first characterized the stable Cl ratios and total sample Cl on a small aliquot removed from the full sample. Using this information, we then prepared the rock samples for 36Cl analysis.
2.2.1 Physical and chemical preparation: all test samples
We prepared all whole-rock samples at the University of New Hampshire Cosmogenic Isotope Lab. Each rock sample had a starting mass of 200–300 g and a thickness of 2–3 cm. We cut some samples with a tabletop rock saw to achieve a uniform thickness. To remove dirt and other contaminants from vesicles, we first crushed samples 19SEAK-12 and 19SEAK-13 to a 1–4 mm grain size. We rinsed these two crushed samples in deionized water and sonicated them in 10 min intervals until no further material could be removed. After the initial crushing and rinsing, we dried the 1–4 mm fractions overnight at 100 °C and then crushed all seven samples to a 125–250 µm grain size. At this stage, we reserved 5–10 g of the 125–250 µm size fraction for trace element analysis, which is necessary to characterize the neutron-moderating properties of the rock for 36Cl exposure age calculations. From the remainder of the 125–250 µm size fraction, we rinsed ∼50 g in deionized water. We leached the samples in a 2 % HNO3 solution for 3 h in heated ultrasonic tanks, rinsed the material in deionized water, and repeated the leaching for a total of two 2 % HNO3 leaches. After the leached samples were dried overnight at 100 °C, we divided the remaining material into two fractions to compare results from standard preparation methods (“A” splits; Fig. 1) and the updated workflow (“B” splits; Fig. 2). In the next sections, we describe the procedures used for the updated workflow in detail.
2.2.2 Characterization of stable Cl ratios: updated workflow
The goal of this step is to characterize stable chlorine ratios () of geologic samples prior to 36Cl chemistry on the full-rock sample. For the test samples, we carried out this process on an ∼1 g aliquot that was removed from the rock sample before beginning the 36Cl extraction (Table 3). After the two 2 % HNO3 etches and subsequent drying at 100 °C, we divided the sample into three fractions using a small spoon (Fig. 2). The first fraction was the ∼1 g aliquot for stable Cl analysis. The second fraction consisted of ∼5–10 g for major element analysis, which is necessary to characterize 36Cl production rates for exposure dating purposes. We reserved the remaining material for 36Cl measurements. To assess the effect of sample homogenization prior to splitting on measured total Cl concentration, we also prepared aliquots of four samples (19SEAK-01, -02, -12, and YGT18-31) using a micro riffle splitter (rather than a spoon) to separate the subsamples from the full rock.
Table 3Laboratory information for test sample measurements and total Cl concentration determinations.
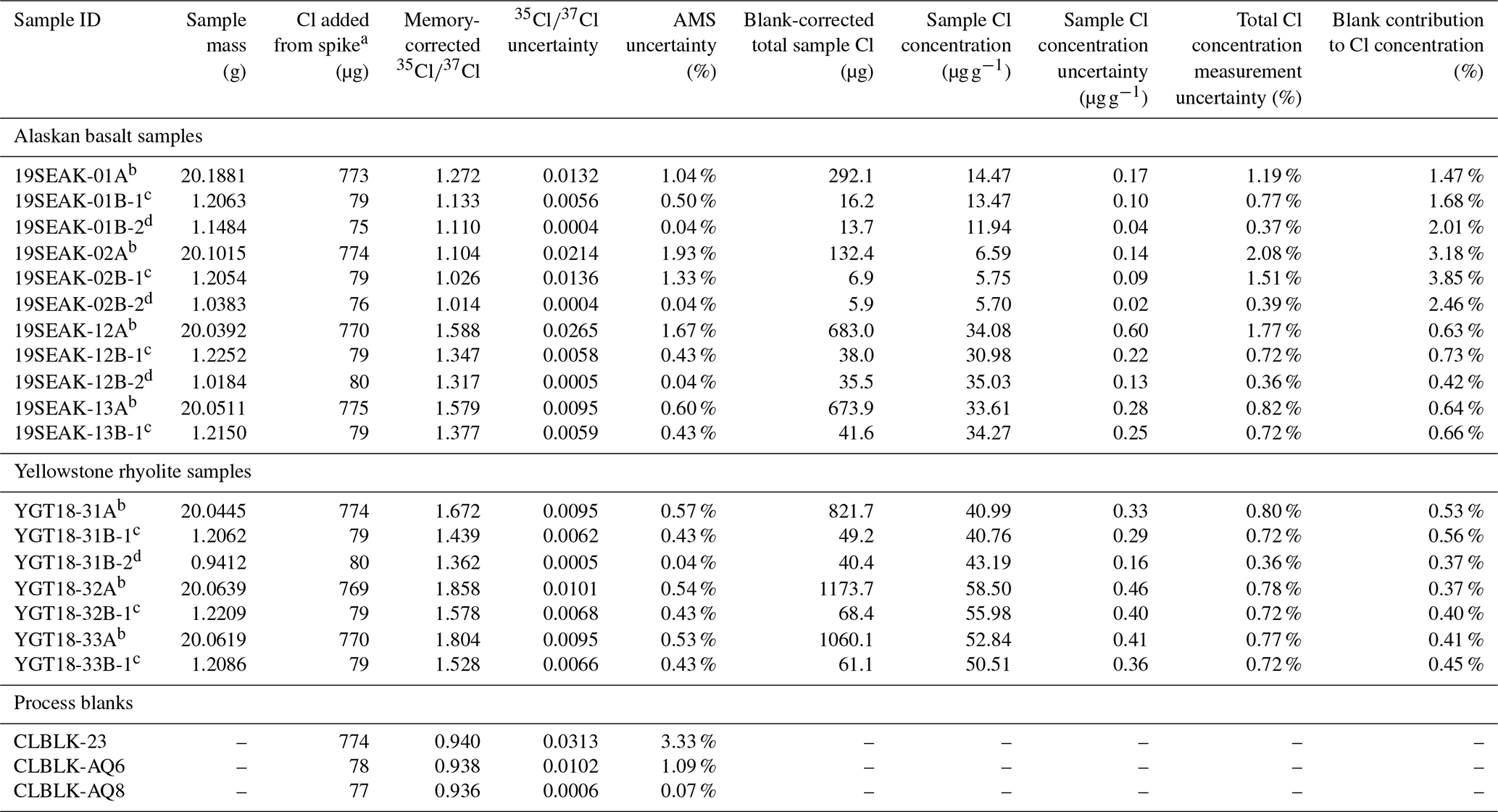
a All samples were spiked with “LLNL Spike A,” which has a of 0.934 and a Cl concentration of 1284 ± 4 µg g−1. b Sample prepared using the standard workflow. Sample Cl concentration corrected using process blank CLBLK-23. c Sample prepared using the updated workflow, without using a micro riffle splitter for homogenization. In addition to the 37Cl-enriched spike, the sample received ∼4000 µg Br from an ∼10 000 µg g−1 NH4Br carrier solution. Sample Cl concentration corrected using process blank CLBLK-AQ6. d Sample prepared using the updated workflow, with homogenization using a micro riffle splitter. In addition to the 37Cl-enriched spike, the sample received ∼4000 µg Br from an ∼10 000 µg g−1 NH4Br carrier solution. Sample Cl concentration corrected using process blank CLBLK-AQ8.
We measured on Ag(Cl, Br) targets prepared using the ∼1 g aliquots of etched rock sample. To prepare the Ag(Cl, Br) targets, we spiked the aliquots with a small amount of LLNL Spike A (Cl concentration = 1284 ± 4 µg g−1, ), totaling ∼75 µg of Cl. We then added ∼4000 µg of bromine to each sample using our NH4Br carrier solution. To promote dissolution, we heated the spiked aliquots to 70 °C for 24–48 h in a solution containing 4 mL of concentrated puriss HF and 6 mL 2 M HNO3 g−1 of sample. After dissolution, we removed fluoride compounds by centrifuging. To precipitate Ag(Cl, Br), we added ∼1 mL of 5 % AgNO3 to the supernatants in low-light conditions and left the solutions to sit in a darkened room. After at least 12 h, we recovered the Ag(Cl, Br) precipitates and transferred them and a small amount of supernatant to centrifuge tubes. We vortexed the solutions then let the precipitates settle for at least 5 min while periodically tapping the bases of the tubes on a table. Then, we centrifuged the tubes to collect the precipitates. All samples underwent two cycles of vortexing, settling, and centrifuging while still in acidic solutions, which facilitated scavenging and flocculation of colloidal Ag(Cl, Br). We then discarded the acidic supernatants and rinsed the precipitates with deionized water. With the Ag(Cl, Br) precipitates now in water, we repeated the vortexing, tapping, and centrifuging steps twice to fully rinse the material and remove residual acids. After discarding the water, we dried the precipitates overnight at 70 °C. Finally, we packed the Ag(Cl, Br) precipitates into nitric-acid-cleaned stainless-steel cathodes for AMS measurement of at CAMS.
We prepared aliquots for measurements in two analytical batches in July 2020 and February 2021, respectively. We used and batch-specific process blanks (Table 3) to calculate total sample chloride through standard isotope dilution methods (Faure and Mensing, 2005). Aliquots for the analytical batch prepared in July 2020 were not separated from the full sample using a micro riffle splitter. The total Cl concentrations for these samples were corrected using process blank CLBLK-AQ6 (measured ± 0.0102). Aliquots for the batch prepared in February 2021 were separated from the full sample after homogenization with a micro riffle splitter. Total sample Cl concentrations for the February 2021 batch were corrected using process blank CLBLK-AQ8 (measured ± 0.0006).
2.2.3 Characterization of cosmogenic 36Cl: updated workflow
We characterized cosmogenic 36Cl concentrations for the updated-workflow (“B”) splits on Ag(Cl, Br) matrices prepared from ≤ 20 g of HNO3-etched rock sample. We carried out the 36Cl extractions in two analytical batches in August 2020 and May 2021. Because we determined the total sample chloride content prior to chemistry on the full sample, we were able to adjust the amount of rock sample and natural-ratio Weeks Island Halite carrier used for 36Cl analyses to ensure consistent total Cl among all targets in each analytical batch while keeping expected for all samples well above laboratory blank values (Table 4). For higher-Cl samples (YGT18-32B and YGT18-33B), no natural-ratio Cl carrier was needed, and the optimal expected and amount of Cl in the Ag(Cl, Br) target (∼750–1000 µg Cl) were achieved by adjusting the amount of rock digested. For low-Cl samples, the optimal expected and total Cl were achieved by adding an appropriate amount of natural-ratio Weeks Island Halite carrier.
Table 4Laboratory information for test sample 36Cl concentration determinations.
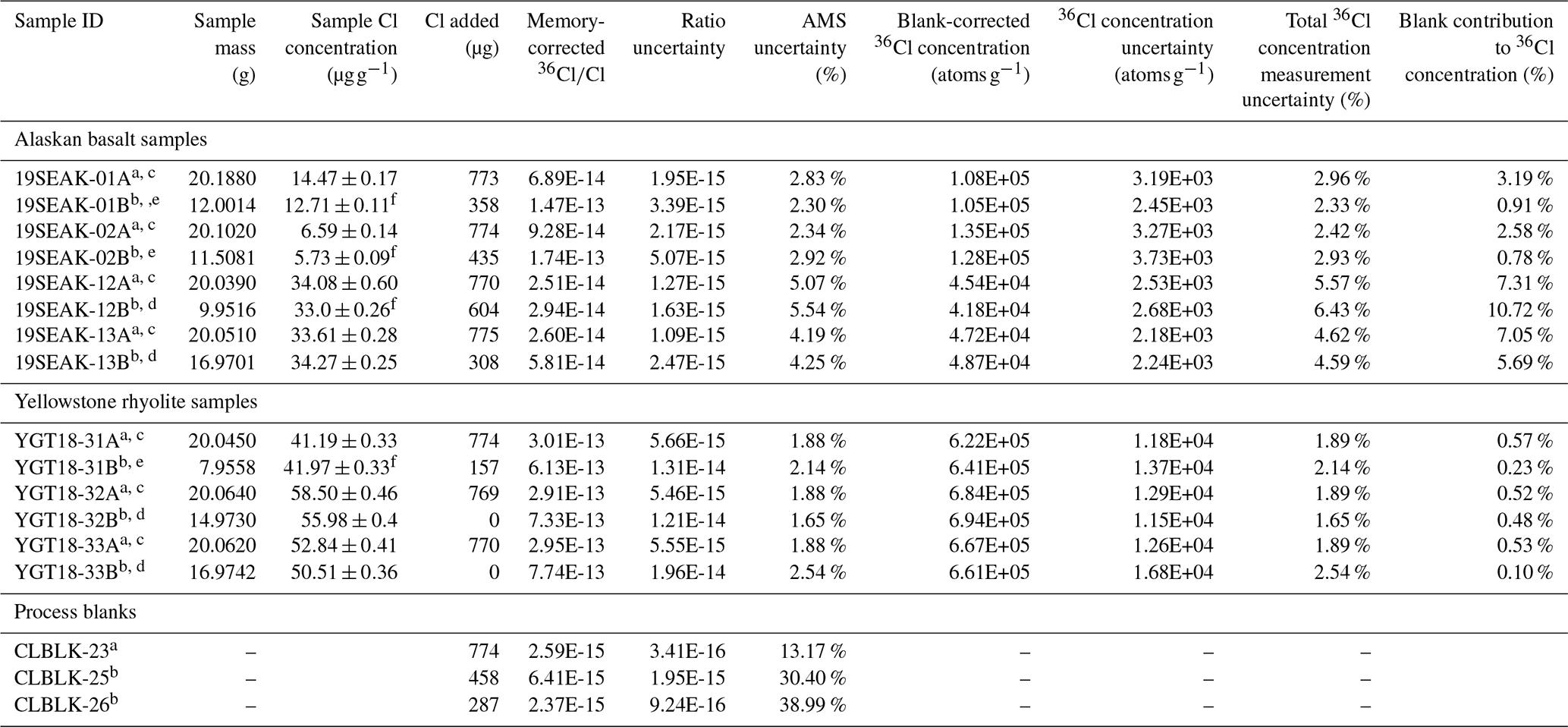
a Sample prepared using the standard workflow. Sample spiked with “LLNL Spike A,” which has a of 0.93 and a Cl concentration of 1285 ± 4 µg g−1. b Sample prepared using the updated workflow. Sample received “Weeks Island Halite carrier” solution, which has a of 3.127 (i.e., natural ratio) and a Cl concentration of 1436 ± 9 µg g−1. Sample also received ∼4000 µg Br from an ∼10 000 µg g−1 NH4Br carrier solution. c Sample 36Cl concentration corrected using process blank CLBLK-23. d Sample 36Cl concentration corrected using process blank CLBLK-25. e Sample 36Cl concentration corrected using process blank CLBLK-26. f Sample Cl concentration used for 36Cl determinations is the average of two stable Cl aliquot measurements (one sample homogenized with a riffle splitter and one sample with no homogenization; Table 3).
Each analytical batch contained a single process blank with Weeks Island Halite carrier and NH4Br carrier. To account for the variable amounts of Weeks Island Halite carrier added to samples within a batch, the process blanks received the average amount of Weeks Island Halite carrier added to the samples in the batch (Table 4). No samples or blanks received 37Cl-enriched spike solution for the 36Cl extraction. All samples and blanks received NH4Br carrier, which served to increase the size of the final precipitate. We added enough NH4Br carrier to each sample within an analytical batch such that the moles of Ag(Cl, Br) were equivalent to the moles of AgCl when precipitating 2000 µg of Cl. Because all samples in an analytical batch had a comparable amount of total Cl after the Weeks Island Halite carrier additions, each sample and process blank in a batch received the same amount of Br carrier.
After the Cl and Br carrier additions, we prepared the Ag(Cl, Br) targets using a modified version of the procedures outlined in Stone et al. (1996) and Licciardi et al. (2008). We dissolved the sample grains in a solution containing 4 mL of concentrated puriss HF and 6 mL 2 M HNO3 g−1 of sample. The solutions were heated to ∼70 °C and left to dissolve over 2 to 3 d. After dissolution, we removed insoluble fluorides through centrifuging. We transferred the supernatants into Teflon beakers and heated them to ∼160 °C before adding ∼1 mL 5 % AgNO3 under low-light conditions. Samples were left to sit in a darkened cabinet for at least 12 h to allow Ag(Cl, Br) to precipitate. We recovered the Ag(Cl, Br) precipitates and then dissolved them in 3.6 M NH3. To remove 36S, an interfering isobar of 36Cl, we separated Cl from S by the addition of saturated BaNO3 to the sample solution. After 2 to 3 d, when BaSO4 crystals had formed in all samples, we transferred the clear supernatant to a new container. We precipitated Ag(Cl, Br) a final time by acidifying the solution with 2 M HNO3 and adding ∼1 mL 5 % AgNO3. After letting the samples sit for at least 12 h in a darkened cabinet, we recovered the Ag(Cl, Br), rinsed the precipitate in deionized water, and dried the material at 70 °C overnight. The precipitates were then sent to CAMS, where they were loaded into stainless-steel targets pre-packed with AgBr and analyzed for .
2.2.4 Isotopic analyses and process blank corrections for 36Cl concentrations
For both the standard-method and the updated-workflow splits, measurements were conducted at LLNL-CAMS. Sample ratios were normalized to KNSTD1600, which has a nominal of and a of (Sharma et al., 1990). To account for laboratory and AMS backgrounds, we corrected all sample 36Cl concentrations using ratios from batch-specific process blanks. The standard-method (“A”) splits were processed in a single analytical batch in November 2019, and their 36Cl concentrations were corrected using a batch-specific blank of 2.59 ± (CLBLK-23; equivalent to 7.18 ± 104 36Cl atoms). The process blank for this batch contained 774 µg of Cl from the 37Cl-enriched spike solution and no Br carrier (Table 4). The updated-workflow (“B”) splits were processed in two analytical batches in August 2020 and May 2021, respectively. The batch processed in August 2020 included four geologic samples and a blank (CLBLK-25) containing 458 µg of Cl from the natural-Cl-ratio Weeks Island Halite carrier and ∼4000 µg of Br from the NH4Br carrier. Sample 36Cl concentrations for the August 2020 batch were corrected using a process blank of 6.41 ± (equivalent to 4.99×104 36Cl atoms). The batch processed in May 2021 included three geologic samples (Table 4) and a process blank (CLBLK-26) that contained 287 µg of Cl from the natural-Cl-ratio Weeks Island Halite carrier and ∼4000 µg of Br from the NH4Br carrier. Sample 36Cl concentrations for the May 2021 batch were corrected using a process blank of 2.37 ± (equivalent to 1.16×104 36Cl atoms).
3.1 Cl dilution series
Measurements of from dilutions of the Wildcat spike and the Weeks Island Halite carrier agree well with expected values over a range of 1.0 to 2.5 (Table 1; Fig. 3), with analytical uncertainties ranging between 0.0079 % and 0.032 %. The close agreement between the measured and expected across the range of values demonstrates that samples analyzed on the low-energy end of the AMS yield accurate and highly precise stable Cl determinations. This result represents a marked improvement over past measurement uncertainties on the high-energy end of the AMS, which average ∼1 %, and demonstrates that low-energy measurements are a favorable alternative to traditional methods.
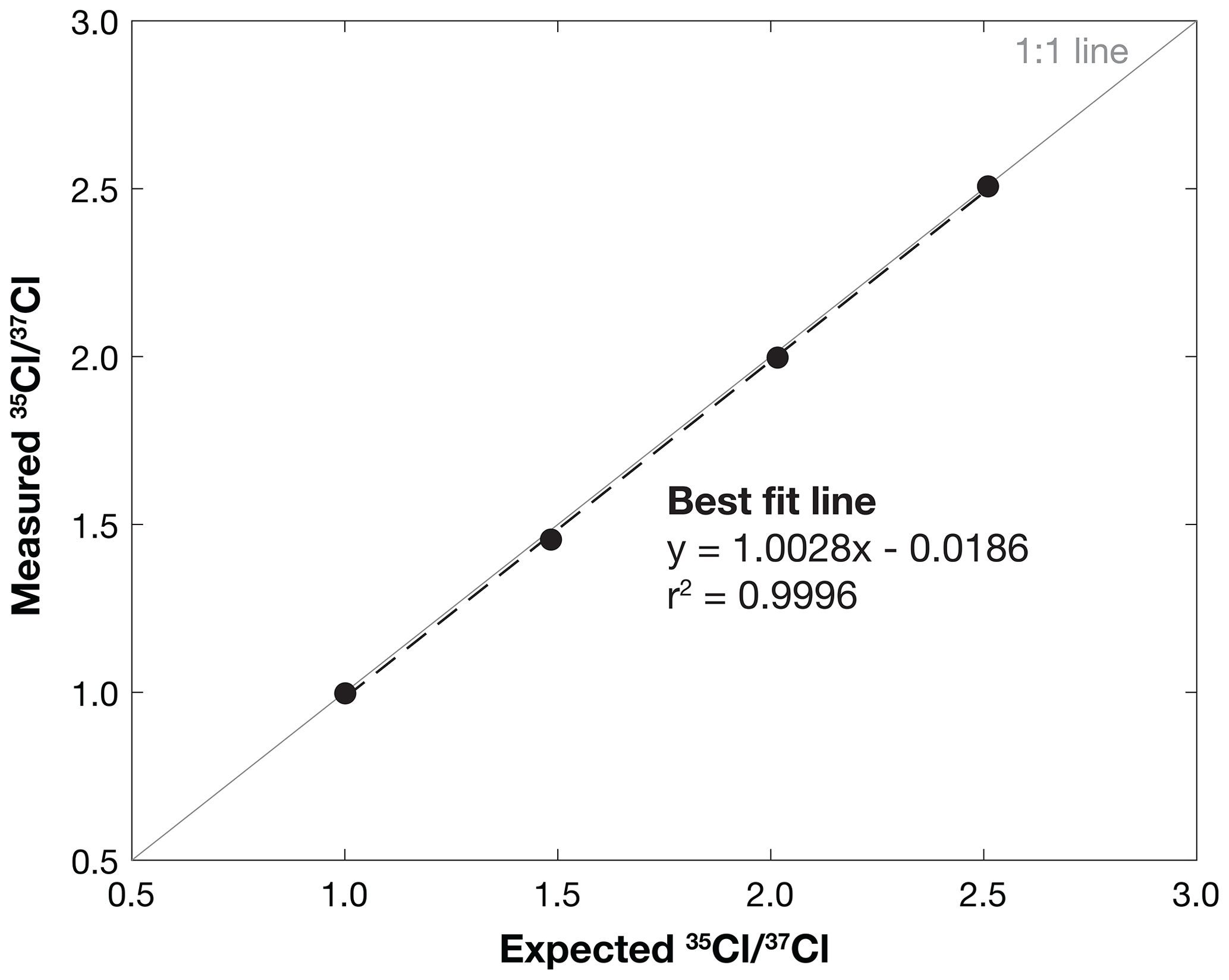
Figure 3Measured vs. expected for dilutions of the UNH Wildcat spike and the Weeks Island Halite carrier. Error bars on measured are too small to be visible on the figure. The solid gray diagonal line shows a 1:1 relationship between expected and measured , and the dotted black line shows the best-fit line between the data points. The equation and the r2 value for the best-fit line are also provided.
Measurements of from dilutions of the Wildcat spike and the NH4Br carrier ranged from 1.012 to 1.036 (Table 2). Counting uncertainties for each target ranged between 0.0045 % and 0.011 %. Although we observe a general trend toward increasing measured in dilutions with less 37Cl-enriched Wildcat spike, isotope dilution calculations reveal that all dilutions contain <2.5 µg Cl g−1 of NH4Br (mean=1.75 ± 0.473 µg g−1); in other words, Cl contamination is negligible in our bromine carrier. Thus, the commercial NH4Br source is suitable for the procedures presented here if samples contain at least 20 µg of total Cl, which is well below the typical target amount for whole-rock and mineral separates analyzed for cosmogenic 36Cl exposure dating. For samples with total Cl masses <20 µg, alternative preparation protocols that do not use a NH4Br carrier (e.g., Anderson et al., 2022) would be necessary. It should be noted, however, that those alternative protocols are not suitable for silicate materials that require digestion in HF during sample dissolution (see Sect. 4, Discussion, below).
3.2 Test samples
We successfully measured and on test samples at levels that were well above process blank values (Tables 3 and 4). For the “A” splits prepared using the standard workflow, errors on measurements ranged from 0.53 % to 1.93 %, resulting in total Cl concentration uncertainties (including process blank corrections) between 0.77 % to 2.08 %. Analytical uncertainty for analyses on the “B” splits prepared using the updated workflow presented here ranged from 0.04 % to 1.33 %, corresponding to total Cl concentration uncertainties (including process blank corrections) from 0.36 % to 1.51 %. These results demonstrate that pre-accelerator measurements of stable Cl isotope ratios can provide higher precision than measurements on the post-accelerator end of the AMS. Blank-corrected total Cl concentrations for test samples varied from ∼6–35 µg g−1 Cl for the Alaskan basalts to ∼41–60 µg g−1 Cl for the Yellowstone rhyolites. (Table 3; Fig. 4). Process blank contributions for samples prepared with both workflows are comparable (Table 3). Total Cl concentrations for the “A” and “B” splits do not overlap at 2σ uncertainty for all samples (Table 3; Fig. 4). For the three Yellowstone rhyolite samples (YGT18-31, YGT18-32, and YGT18-33), the total Cl determinations for the “A” and “B” splits are within 5 % of one another. For the four Alaskan basalt samples (19SEAK-01, 19SEAK-02, 19SEAK-12, and 19SEAK-13), the difference in total Cl concentration between the “A” and “B” splits ranges from 2 % to 13 %. This scatter is likely due to small-scale compositional heterogeneity in rock sample composition or dissolution.
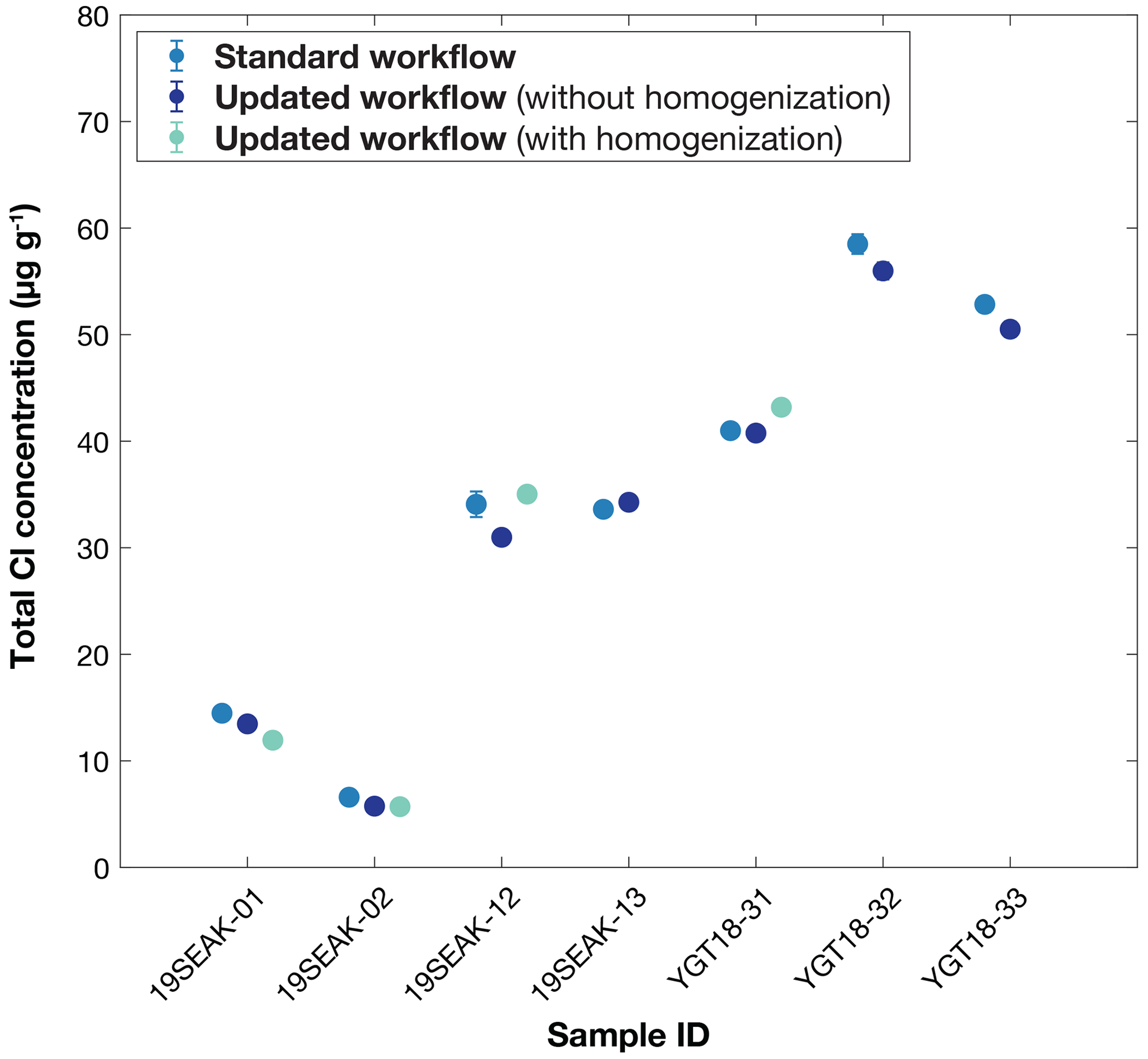
Figure 4Comparison of total Cl concentrations (µg g−1) for samples measured using the standard-workflow “A” splits (blue dots; all samples), the updated-workflow “B-1” splits with aliquots separated without homogenization (purple dots; all samples), and the updated-workflow “B-2” splits with aliquots separated after homogenization with a riffle splitter (green dots; four samples only). With the exception of 19SEAK-12A and YGT18–32A, the total uncertainties for each sample (Table 3) are smaller than the symbol size. For 19SEAK12A and YGT18–32A, 2σ uncertainty is shown as a vertical line.
36Cl concentrations for the standard-method “A” splits ranged from 4.54×104 to 6.84×105 atoms g−1, with total measurement uncertainties (including process blank corrections) between 1.89 % and 5.57 % (Table 4). For the updated-workflow “B” splits, 36Cl concentrations ranged from 4.18×104 to 6.94×105 atoms g−1; total measurement uncertainties ranged from 1.65 % to 6.43 %. Uncertainties on measurements are higher than on measurements, which is not surprising given the much lower ratios measured for . 36Cl concentrations for all sample pairs agree within 1σ measurement uncertainty. There are no systematic variations in 36Cl concentration between the two preparation workflows (Fig. 5), and process blank contributions to 36Cl concentrations are also comparable for all samples (Table 4). This set of measurements demonstrates that the 36Cl analyses for each preparation method provide equivalent results within uncertainty.
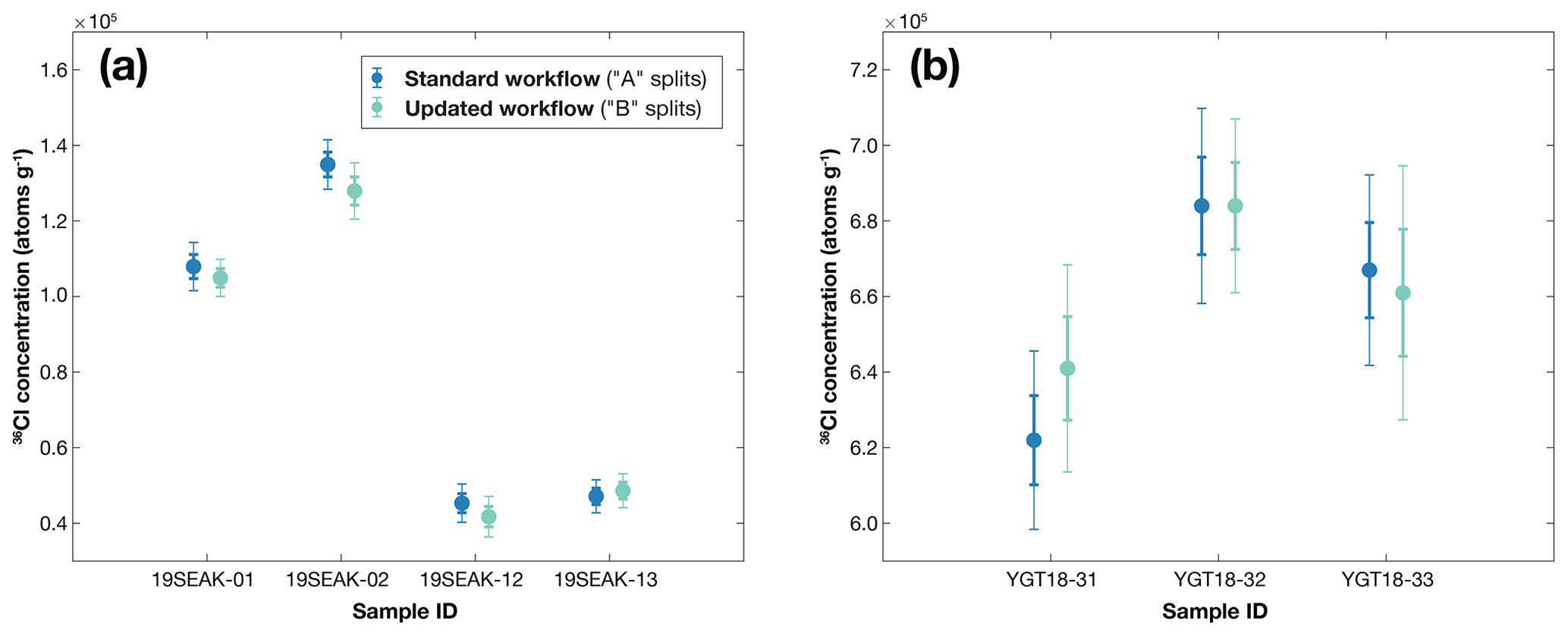
Figure 5Comparison of blank-corrected 36Cl concentrations (atoms g−1) for the geologic test samples, (a) Alaskan basalt and (b) Yellowstone rhyolite, prepared using the standard workflow (“A” splits; blue dots) and the updated workflow (“B” splits; green dots) presented here. Measurement uncertainties for each sample are shown at 1σ (thick vertical lines) and 2σ (thin vertical lines). Although measurement uncertainties on 36Cl concentrations for the three YGT18 samples appear visually larger than those of the 19SEAK samples, percent uncertainties on both sets of samples are comparable (Table 4). For ease of comparison, the range of the y axis in both panels is 1.4×105 atoms g−1.
The cosmogenic chlorine workflow presented here offers substantial advantages over conventional protocols across all levels of analysis, spanning from laboratory preparation to AMS measurement. Measurement of on the low-energy end of the AMS line with consistent and substantially smaller total Cl loads in all targets results in total uncertainties on measurements of <1 % and often <0.5 %. Rock-sample chlorine concentrations can thus be determined with high precision, which will reduce external uncertainties on calculated surface exposure ages. By characterizing and total Cl prior to 36Cl measurement, rock samples with high Cl concentrations can also be identified and screened at an earlier stage of analysis. This is important because high native Cl concentrations in rocks can result in exposure ages with comparably larger external uncertainties due to the greater uncertainty in the 36Cl production rate from thermal and epithermal neutron capture on 35Cl (Marrero et al., 2016). Depending on the desired exposure age precision, these high native Cl samples may be excluded from further analysis. Furthermore, the aliquot method presented here uses only ∼1 g of etched sample to obtain total Cl concentrations (Stone, 2001). However, we note that this method can be applied to substantially less material than 1 g depending on a sample's total Cl concentration, aliquot homogeneity, and the need for high precision in the Cl concentration measurement. We chose to use 1 g of etched sample to help ensure that the stable Cl aliquot was representative of the whole sample and because we suspected some samples could have exceptionally low Cl concentrations such that 1 g would be large enough to characterize Cl at levels where the 35Cl(n,γ)36Cl reaction becomes a significant contributor to total 36Cl production. For the method presented here, a sample size equivalent to 5 µg Cl is likely sufficient, indicating that many of our samples could have been analyzed at high precision using only ∼0.1 g of material. This is substantially less than the amount required for total Cl determinations by other techniques such as XRF, which can consume >10 g of material. The small amount of material required for total Cl measurements is thus a key advantage when sample sizes are limited (e.g., when analyzing mineral separates). Finally, and perhaps most importantly for cosmogenic chlorine extraction laboratories, our workflow reduces the use of isotopically enriched chlorine spike by up to 95 % compared to conventional methods, which will considerably extend the lifespan of existing laboratory supplies.
While our laboratory protocols offer an improvement over conventional cosmogenic chlorine preparation methods, there is room for further refinement. For example, Anderson et al. (2022) presented an innovative approach wherein AgCl targets were prepared within a niobium matrix, resulting in successful measurement of on targets with Cl masses as low as 1 µg; they also observed a substantially reduced source memory compared to targets bulked with AgBr. However, a key distinction between their methodology and the one outlined here is that, unlike our silicate samples, the aqueous Cl samples of Anderson et al. did not require the use of HF in the dissolution process. Given niobium's solubility in HF acid, employing this method on silicate samples requires thorough rinsing to eliminate residual acid from AgCl precipitates before introducing niobium powder. HF remaining in the AgCl precipitates can also react to form fluoride compounds with an atomic mass of 35 (e.g., 16O19F), which can interfere with the measurement of . Thus, removing all HF before adding niobium to AgCl precipitates is critical. On the other hand, AgCl is mildly water soluble (∼2 µg mL−1), and extensive rinsing may reduce the AgCl yield. Therefore, the challenge is to rinse samples thoroughly enough to remove all traces of HF while still maintaining a high AgCl yield. We have attempted this process with silicate-derived AgCl precipitates that contain ∼50 µg of total Cl, but consistently and accurately measuring on such samples has proven difficult, likely due to both yield loss during the procedure and residual HF remaining in some targets even after thorough rinsing. Refining these procedures is the focus of ongoing experimentation. Nevertheless, our findings unequivocally show that on silicate samples can be measured with high precision on Ag(Cl, Br) when the total chlorine mass in each target exceeds 50 µg. Thus, while there are possibilities for improvement, we are confident that the procedures presented herein can be deployed with little modification in cosmogenic chlorine laboratories and AMS facilities.
We note that adopting these procedures would slightly increase the time and lab resources required to extract 36Cl from rock samples. For example, although can be measured on the low-energy end of the AMS line, which does not require a “full-scale” AMS run, sample shipment and data processing would add some time between sample preparation and the receipt of final 36Cl concentrations. The separate extraction adds approximately 3 to 4 d of laboratory work to the 36Cl extraction timeline and also requires small amounts of reagents such as HF, HNO3, and AgNO3. However, in cases where the total sample Cl is high, 36Cl extractions can be carried out on smaller masses of milled rock; thus the total volume of acid used in both the and 36Cl extractions may be smaller than what was required to dissolve ∼20 g of milled rock using the standard workflow. Furthermore, having knowledge of Cl concentration in advance of the 36Cl analysis allows the screening out of samples with high Cl content that may be undesirable for certain applications. In such cases, this workflow saves both time and resources that would otherwise be wasted on less useful 36Cl data. In terms of the analytical cost, presently at the CAMS facility there is no cost difference between analyzing on a separate, earlier target and analyzing both and on the same target during a single AMS run. However, for a measurement used to screen samples (that is, when no later analysis is performed), the analysis cost is ∼ one-seventh that of the -inclusive analysis such that significant cost savings can be achieved when sample screening is prudent.
To implement the preparation and measurement workflow described above, we suggest three key practices. Firstly, we recommend using a micro riffle splitter to separate subsamples for , , and major element analyses from the full-rock sample. The homogenization step does not appear to be strictly necessary for the two lithologies we tested here (basalt and rhyolite; Fig. 4). We speculate that these fine-grained igneous rocks are already quite compositionally homogenous due to the scarcity or lack of phenocrysts and the high percentage of groundmass; therefore the milled grains do not separate much by composition after milling and during storage in plastic bags. However, we do encourage the use of a micro riffle splitter or another homogenization method (e.g., the “cone and quarter” technique) for coarse-grained lithologies that may contain monomineralic grains when crushed to the 250–125 µm size fraction we recommend here, as we suspect these rocks may be more susceptible to separation by composition during storage. Secondly, we stress the importance of exercising caution when cleaning stainless-steel AMS cathodes prior to sample loading. Early experiments indicated that the commercial laboratory soaps we used to clean our AMS cathodes contain chlorine that is not removed from cathode surfaces by thorough rinsing with deionized water. Therefore, we suggest that laboratory users soak AMS cathodes in a weak (∼1 %) nitric acid solution after cleaning with laboratory soap. This step is essential to eliminate residual natural-ratio chlorine that may contaminate the cathode surfaces and erroneously raise measured on spiked samples. Thirdly, if samples are bulked with NH4Br carrier, we recommend that laboratory users prepare “matrix blanks” of AgBr (with no added Cl) for measurement, which will allow accurate ion-source memory corrections for unknown samples. Consistency in target matrices across a stable Cl analysis minimizes source memory and improves ion beam stability, contributing to more accurate and reproducible experimental outcomes.
The workflow presented here uses a 37Cl-enriched spike solution for the measurements, but it is also suitable for use with a 35Cl-enriched spike so long as the solution is sufficiently enriched relative to the natural of 3.127. In general, solutions enriched in 35Cl are more readily available than 37Cl-enriched solutions, so this may be an attractive alternative for laboratories that cannot acquire a 37Cl-enriched spike.
Our workflow for extracting and measuring chlorine in silicate rocks improves upon standard preparation methods in several key ways. After crushing the rock and cleaning the mineral surfaces with dilute HNO3, we characterize stable Cl ratios on an aliquot of rock of up to ∼1 g removed from the full sample. is then measured on the low-energy beam line of the AMS accelerator, allowing us to quickly determine total chlorine loads while minimizing source memory and reducing the amount of 37Cl-enriched carrier solution used per sample by up to 95 % compared to traditional methods. With data in hand, we then extract Cl from rock samples for measurements and bulk the AMS target material with bromine and/or natural-ratio chlorine solutions without adding a 37Cl-enriched spike. Experiments on seven geologic test samples reveal that the workflow presented here yields comparable or, in the case of the measurements, improved results over the traditional workflow. Most notably, by measuring on an ∼1 g aliquot rather than on a 20 g sample, the updated preparation methods use substantially less isotopically enriched spike solution than standard methods (∼50–75 µg Cl versus ∼750–1000 µg Cl). With lowered spike solution requirements, researchers can analyze many more samples using their remaining laboratory resources. Chlorine extraction laboratories will also be able to maintain control over the total chlorine content within and across analytical batches. Finally, in comparison to the standard 36Cl workflow, our method can identify samples with elevated native Cl concentrations at an earlier stage of laboratory work, which can help researchers determine which of their rock samples should be prioritized for 36Cl analyses. Our hope is that these procedures will supplement existing laboratory and AMS workflows for cosmogenic Cl and enhance the effectiveness of 36Cl dating for a variety of geologic applications.
All data are presented in Tables 1–4 of this technical note.
AJL and JML carried out the laboratory work. AJH and TSA made the and measurements. AJL wrote the first draft of the paper, and all authors contributed to experimental design, data analysis, and article review and editing.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors thank Shasta Marrero and Irene Schimmelpfennig for their thoughtful and constructive reviews. We also thank John Stone and Keith Fifield for helpful discussions.
This work has been supported in part by LLNL under contract no. DE-AC52-07NA27344 (LLNL-JRNL-861073).
This paper was edited by Hella Wittmann and reviewed by Shasta Marrero and Irene Schimmelpfennig.
Anderson, T. S., Hidy, A. J., Boyce, J. W., McCubbin, F. M., Tumey, S., Dudley, J. M., Haney, N. C., Bardoux, G., and Bonifacie, M.: Development towards stable chlorine isotope measurements of astromaterials using the modified Middleton source of an accelerator mass spectrometer, Int. J. Mass Spectrom., 477, 116849, https://doi.org/10.1016/J.IJMS.2022.116849, 2022.
Arnold, M., Merchel, S., Bourlès, D. L., Braucher, R., Benedetti, L., Finkel, R. C., Aumaître, G., Gottdang, A., and Klein, M.: The French accelerator mass spectrometry facility ASTER: improved performance and developments, Nucl. Instrum. Meth. B, 268, 1954–1959, 2010.
Barth, A. M., Marcott, S. A., Licciardi, J. M., and Shakun, J. D.: Deglacial Thinning of the Laurentide Ice Sheet in the Adirondack Mountains, New York, USA, Revealed by 36Cl Exposure Dating, Paleoceanogr. Paleoclimatol., 34, 946–953, https://doi.org/10.1029/2018PA003477, 2019.
Ben-Asher, M., Haviv, I., Crouvi, O., Roering, J. J., and Matmon, A.: The convexity of carbonate hilltops: 36Cl constraints on denudation and chemical weathering rates and implications for hillslope curvature, GSA Bulletin, 133, 1930–1946, https://doi.org/10.1130/B35658.1, 2021.
Benedetti, L., Finkel, R., Papanastassiou, D., King, G., Armijo, R., Ryerson, F., Farber, D., and Flerit, F.: Post-glacial slip history of the Sparta fault (Greece) determined by 36Cl cosmogenic dating: Evidence for non-periodic earthquakes, Geophys. Res. Lett., 29, 87-1–87-4, https://doi.org/10.1029/2001GL014510, 2002.
Eberlein, G. D., Churkin, M., Carter, C., Berg, H. C., and Ovenshine, A. T.: Geology of the Craig quadrangle, Alaska, US Geological Survey, https://doi.org/10.3133/ofr8391, 1983.
Faure, G. and Mensing, T. M.: Isotopes: Principles and Applications, 3rd Edn., John Wiley and Sons, Hoboken, New Jersey, ISBN 978-0-471-38437-3, 2005.
Finkel, R., Arnold, M., Aumaître, G., Benedetti, L., Bourlès, D., Keddadouche, K., and Merchel, S.: Improved 36Cl performance at the ASTER HVE 5 MV accelerator mass spectrometer national facility, Nucl. Instrum. Meth. B, 294, 121–125, 2013.
Gosse, J. C. and Phillips, F. M.: Terrestrial in situ cosmogenic nuclides: Theory and application, Quaternary Sci. Rev., 20, 1475–1560, https://doi.org/10.1016/S0277-3791(00)00171-2, 2001.
Ivy-Ochs, S., Poschinger, A. V., Synal, H.-A., and Maisch, M.: Surface exposure dating of the Flims landslide, Graubünden, Switzerland, Geomorphology, 103, 104–112, 2009.
Kozaci, O., Dolan, J., Finkel, R., and Hartleb, R.: Late Holocene slip rate for the North Anatolian fault, Turkey, from cosmogenic 36Cl geochronology: Implications for the constancy of fault loading and strain release rates, Geology, 35, 867–870, 2007.
Licciardi, J. M. and Pierce, K. L.: History and dynamics of the Greater Yellowstone Glacial System during the last two glaciations, Quaternary Sci. Rev., 200, 1–33, 2018.
Licciardi, J. M., Denoncourt, C. L., and Finkel, R. C.: Cosmogenic 36Cl production rates from Ca spallation in Iceland, Earth Planet. Sc. Lett., 267, 365–377, https://doi.org/10.1016/J.EPSL.2007.11.036, 2008.
Marrero, S. M., Phillips, F. M., Caffee, M. W., and Gosse, J. C.: CRONUS-Earth cosmogenic 36Cl calibration, Quat. Geochronol., 31, 199–219, 2016.
Marrero, S. M., Hein, A. S., Naylor, M., Attal, M., Shanks, R., Winter, K., Woodward, J., Dunning, S., Westoby, M., and Sugden, D.: Controls on subaerial erosion rates in Antarctica, Earth Planet. Sc. Lett., 501, 56–66, https://doi.org/10.1016/J.EPSL.2018.08.018, 2018.
Mitchell, S. G., Matmon, A., Bierman, P. R., Enzel, Y., Caffee, M., and Rizzo, D.: Displacement history of a limestone normal fault scarp, northern Israel, from cosmogenic 36Cl, J. Geophys. Res.-Sol. Ea., 106, 4247–4264, https://doi.org/10.1029/2000JB900373, 2001.
Pánek, T., Lenart, J., Hradecký, J., Hercman, H., Braucher, R., Šilhán, K., and Škarpich, V.: Coastal cliffs, rock-slope failures and Late Quaternary transgressions of the Black Sea along southern Crimea, Quaternary Sci. Rev., 181, 76–92, 2018.
Parmelee, D. E. F., Kyle, P. R., Kurz, M. D., Marrero, S. M., and Phillips, F. M.: A new Holocene eruptive history of Erebus volcano, Antarctica using cosmogenic 3He and 36Cl exposure ages, Quat. Geochronol., 30, 114–131, 2015.
Phillips, F. M., Zreda, M. G., Gosse, J. C., Klein, J., Evenson, E. B., Hall, R. D., Chadwick, O. A., and Sharma, P.: Cosmogenic 36Cl and 10Be ages of Quaternary glacial and fluvial deposits of the Wind River Range, Wyoming, Geol. Soc. Am. Bull., 109, 1453–1463, 1997.
Price, B. N., Stansell, N. D., Fernández, A., Licciardi, J. M., Lesnek, A. J., Muñoz, A., Sorensen, M. K., Jaque Castillo, E., Shutkin, T., Ciocca, I., and Galilea, I.: Chlorine-36 Surface Exposure Dating of Late Holocene Moraines and Glacial Mass Balance Modeling, Monte Sierra Nevada, South-Central Chilean Andes (38° S), Front. Earth Sci., 10, 848652, https://doi.org/10.3389/feart.2022.848652, 2022.
Riehle, J. R., Brew, D. A., and Lanphere, M. A.: Geologic map of the Mount Edgecumbe volcanic field, Kruzof Island, southeastern Alaska, U.S. Geological Survey Miscellaneous Investigations Series Map 1983, 1 sheet, scale 1:63, 360, 1989.
Robertson, J., Meschis, M., Roberts, G. P., Ganas, A., and Gheorghiu, D. M.: Temporally constant Quaternary uplift rates and their relationship with extensional upper-plate faults in south Crete (Greece), constrained with 36Cl cosmogenic exposure dating, Tectonics, 38, 1189–1222, 2019.
Schlagenhauf, A., Manighetti, I., Benedetti, L., Gaudemer, Y., Finkel, R., Malavieille, J., and Pou, K.: Earthquake supercycles in Central Italy, inferred from 36Cl exposure dating, Earth Planet. Sc. Lett., 307, 487–500, https://doi.org/10.1016/J.EPSL.2011.05.022, 2011.
Sharma, P., Kubik, P. W., Fehn, U., Gove, H. E., Nishiizumi, K., and Elmore, D.: Development of 36Cl standards for AMS, Nucl. Instrum. Meth. B, 52, 410–415, https://doi.org/10.1016/0168-583X(90)90447-3, 1990.
Singer, B. S., Le Mével, H., Licciardi, J. M., Córdova, L., Tikoff, B., Garibaldi, N., Andersen, N. L., Diefenbach, A. K., and Feigl, K. L.: Geomorphic expression of rapid Holocene silicic magma reservoir growth beneath Laguna del Maule, Chile, Sci. Adv., 4, eaat1513, https://doi.org/10.1126/sciadv.aat1513, 2018.
Small, D., Rinterknecht, V., Austin, W. E. N., Bates, R., Benn, D. I., Scourse, J. D., Bourlès, D. L., Hibbert, F. D., and ASTER Team: Implications of 36Cl exposure ages from Skye, northwest Scotland for the timing of ice stream deglaciation and deglacial ice dynamics, Quaternary Sci. Rev., 150, 130–145, 2016.
Stone, J. O.: University of Washington Cosmogenic Isotope Laboratory Procedures, http://depts.washington.edu/cosmolab/chem.shtml (last access: 30 May 2024), 2001.
Stone, J. O., Allan, G. L., Fifield, L. K., and Cresswell, R. G.: Cosmogenic chlorine-36 from calcium spallation, Geochim. Cosmochim. Ac., 60, 679–692, 1996.
Walcott, C. K., Briner, J. P., Baichtal, J. F., Lesnek, A. J., and Licciardi, J. M.: Cosmogenic ages indicate no MIS 2 refugia in the Alexander Archipelago, Alaska, Geochronology, 4, 191–211, https://doi.org/10.5194/gchron-4-191-2022, 2022.
Wilcken, K. M., Freeman, S., Schnabel, C., Binnie, S. A., Xu, S., and Phillips, R. J.: 36Cl accelerator mass spectrometry with a bespoke instrument, Nucl. Instrum. Meth. B, 294, 107–114, 2013.
Zerathe, S., Lebourg, T., Braucher, R., and Bourlès, D.: Mid-Holocene cluster of large-scale landslides revealed in the Southwestern Alps by 36Cl dating. Insight on an Alpine-scale landslide activity, Quaternary Sci. Rev., 90, 106–127, 2014.